Will Future Medicine Transplant Organelles Instead of Organs?
- Details
- Published on 20 January 2026



Mitochondrial transplantation is emerging as a potential new strategy to restore cellular energy and repair damaged tissues.
For decades, medicine has replaced failing organs through transplantation. But a new scientific idea is beginning to reshape biomedical thinking: what if we could repair cells by replacing their organelles instead of entire organs?
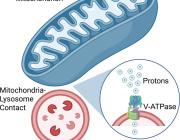
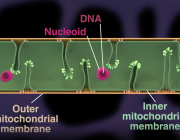
At the center of this emerging concept are mitochondria, the organelles responsible for producing cellular energy and regulating metabolism, oxidative stress, and cell survival. Mitochondrial dysfunction is now recognized as a central component of many diseases, including neurodegeneration, metabolic disorders, ischemic injury, and inflammatory conditions.
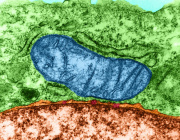
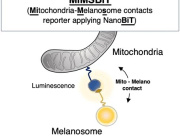
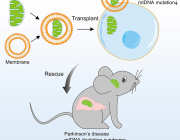
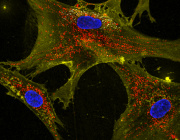
Recent experimental work has begun to clarify how cells interact with transplanted mitochondria. Researchers studying mesenchymal stromal cells demonstrated that isolated mitochondria can be actively internalized by recipient cells, rather than remaining outside the cell.
Once internalized, these mitochondria remain metabolically active and enhance key cellular functions. The study showed increases in oxygen consumption, ATP production, and resistance to oxidative stress, indicating improved cellular bioenergetics.
Importantly, mitochondrial uptake occurs through energy-dependent endocytic pathways, including dynamin-mediated and lipid-raft–associated mechanisms. These findings suggest that mitochondrial incorporation is an active biological process rather than a passive event.
A Strategic Topic for Future Mitochondrial Medicine
The concept of mitochondrial transplantation reflects a broader transformation in biomedical thinking: mitochondria are no longer viewed only as metabolic organelles but as therapeutic targets and potential therapeutic agents.
For this reason, mitochondrial transfer and mitochondrial-based therapies will be one of the strategic themes discussed at the upcoming Targeting Mitochondria 2026 meeting, organized by the World Mitochondria Society in Berlin.
The meeting will explore several emerging directions in mitochondrial medicine, including:
- Mitochondrial transplantation and organelle therapy
- Mitochondrial communication with microbiota and immune systems
- Extracellular vesicles as carriers of mitochondrial signals
- Metabolic resilience and aging
- Mitochondrial dysfunction in neurodegeneration and chronic disease
Understanding how mitochondria move between cells, integrate into recipient tissues, and influence cellular metabolism may redefine how medicine approaches diseases driven by energy failure.
Implications for Medicine
Because mitochondrial dysfunction contributes to numerous diseases, mitochondrial transplantation could potentially impact several fields:
- Neurodegenerative diseases such as Alzheimer’s and Parkinson’s disease
- Stroke and ischemia–reperfusion injury
- Metabolic diseases
- Inflammatory and immune disorders
In neuroscience, experimental studies are already exploring whether delivering mitochondria to damaged neurons could protect brain cells and improve recovery after injury.
Toward Organelle-Based Medicine
Traditional therapies target molecules or genes. Mitochondrial transplantation proposes something fundamentally different: restoring cellular function by supplying intact bioenergetic organelles.
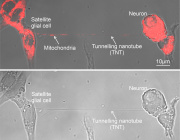
Interestingly, biology already supports this possibility. Cells can naturally exchange mitochondria through tunneling nanotubes and extracellular vesicles, suggesting that organelle transfer may be part of natural repair mechanisms.
If these processes can be controlled and scaled, medicine may eventually move toward a new paradigm: organelle-based therapies.
The Questions That Will Shape the Field
Despite its promise, mitochondrial transplantation remains an emerging concept. Several scientific challenges remain:
- How efficiently can transplanted mitochondria integrate into tissues?
- How long do transplanted mitochondria remain functional?
- Can mitochondrial delivery become safe, scalable, and standardized?
- What regulatory frameworks will govern organelle-based therapies?
Answering these questions will determine whether mitochondrial transplantation becomes a cornerstone of regenerative medicine.
If successful, future therapies might not only repair genes or proteins.
They might restore the energy systems of the cell itself.
References
Kanai, M., Goto, M., Itakura, S. et al. Uptake mechanisms and functions of isolated mitochondria in mesenchymal stromal cells. Sci Rep15, 44799 (2025). https://doi.org/10.1038/s41598-025-28494-5
A new driver of inflammation in aging: Mitochondrial RNA leakage
- Details
- Published on 14 January 2026
A major new study authored by Stella Victorelli and Madeline Eppard and led by Joao Passos, of Mayo Clinic, uncovers a previously unknown mechanism driving chronic inflammation in aging.
The research indicates that mitochondrial RNA (mtRNA) leaking into the cytosol of senescent cells acts as a key trigger for the senescence-associated secretory phenotype (SASP), a significant factor contributing to tissue dysfunction and age-related diseases.
What the study found:
- Senescent cells accumulate mtRNA in the cytosol, where it activates innate immune RNA sensors RIG-I and MDA5, leading to Mitochondrial Antiviral Signaling protein (MAVS) aggregation and inflammatory signaling.
- The study identifies BAX and BAK-dependent mitochondrial membrane permeabilization as the mechanism allowing mtRNA leakage. Genetic deletion of BAX/BAK suppresses SASP both in vitro and in vivo.
- In a mouse model of metabolic dysfunction–associated steatohepatitis (MASH), inhibiting this mtRNA-RNA sensing axis reduced liver inflammation and fibrosis markers, demonstrating physiological relevance.
Why it matters:
This work expands the aging paradigm beyond mitochondrial DNA by identifying mtRNA as a potent endogenous danger signal. It reveals a new mitochondria-to-cytosol signaling axis that fuels chronic inflammation in aging and age-related disease. It highlights RNA sensing, MAVS, and mitochondrial permeability as promising therapeutic targets to mitigate inflammaging without eliminating senescent cells.
References:
Victorelli, S., Eppard, M., Martini, H. et al. Mitochondrial RNA cytosolic leakage drives the SASP. Nat Commun16, 10992 (2025). https://doi.org/10.1038/s41467-025-66159-z
Exercise Does More Than Move Muscles It Moves Mitochondria
- Details
- Published on 23 December 2025
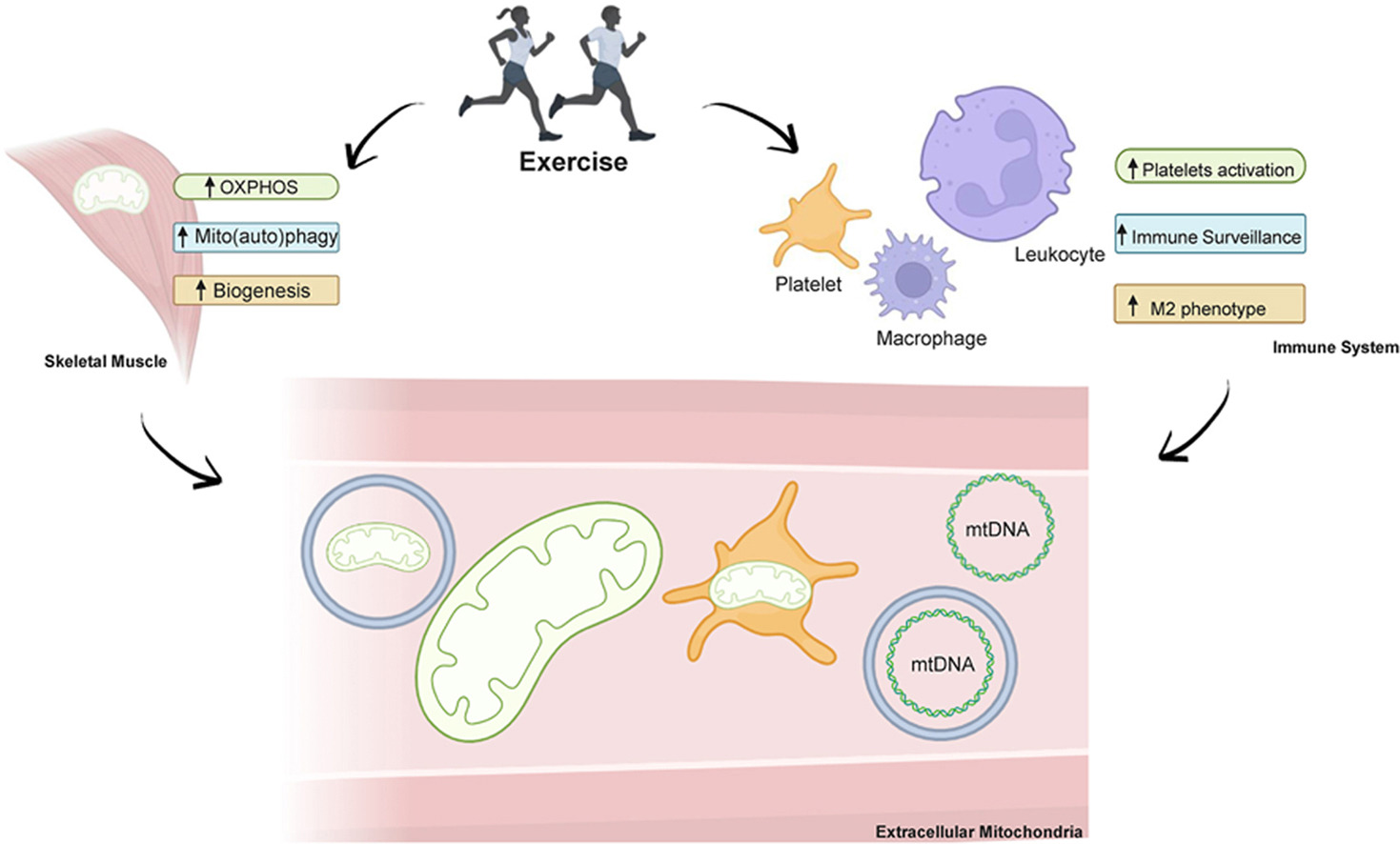
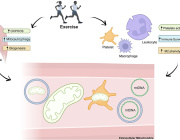
For decades, mitochondria have been described as the cell’s powerhouses - essential for energy production, yet largely silent and confined inside the cell. A new scientific review invites us to rethink that view. It suggests that exercise may do something far more profound: set mitochondria in motion and turn them into systemic messengers of health.
The review explores growing evidence that physical activity can lead to the presence of mitochondria, or mitochondrial components, in the bloodstream. These extracellular mitochondria appear in different forms - sometimes free, sometimes associated with vesicles or blood elements - pointing to a complex and still largely unexplored mode of biological communication triggered by movement.
Exercise has long been known to reshape mitochondria within muscles, the heart, and the brain. What is new here is the shift in perspective. Instead of focusing only on what happens inside cells, the authors look at what may happen between organs. The idea that mitochondria could circulate and carry signals fits with a more modern, systemic understanding of physiology, where health emerges from coordination rather than isolation.
Until now, circulating mitochondrial material has often been viewed through the lens of stress or damage, particularly mitochondrial DNA. This review challenges that narrow interpretation. It raises the possibility that extracellular mitochondria are not just by-products, but active participants in biological signaling, potentially influencing inflammation, metabolism, immune balance, and adaptation to physical effort.
Many questions remain unanswered. Where do these circulating mitochondria come from? Are they functional? Do they signal resilience, adaptation, or cellular strain - or all of these depending on context? What is clear is that this field is still in its early stages, and its implications could be far-reaching.
What makes this work especially timely is its resonance with a broader shift in medicine. Health is no longer seen as static, nor disease as purely organ-based. Instead, attention is turning toward dynamic processes, communication networks, and resilience mechanisms. In this context, mitochondria are no longer just engines of energy. They become messengers, carrying information about physiological state and adaptation.
Seen through this lens, exercise is not simply about movement, performance, or calorie expenditure. It may be a way of activating a circulating mitochondrial dialogue, coordinating responses across tissues and shaping long-term systemic health.
In the spirit of WMS 2026, this review does more than add a new mechanism. It opens a new way of thinking: What if movement speaks the language of mitochondria and mitochondria carry that message throughout the body?
Mitochondrial Dysfunction: The “Mother” of All Hallmarks of Aging
- Details
- Published on 23 December 2025
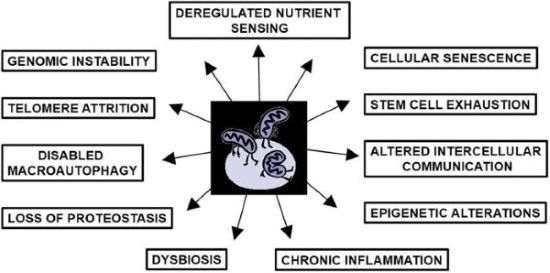
We are pleased to highlight a new review by Prof. Volkmar Weissig, President of the World Mitochondria Society: “Mitochondrial dysfunction as the ‘mother’ of all hallmarks of aging” , published in Journal of Mitochondria, Plastids and Endosymbiosis.
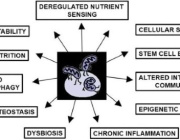
This comprehensive review revisits the role of mitochondria in aging, tracing back to Denham Harman’s Mitochondrial Free Radical Theory of Aging. While mitochondrial contributions have often been underrepresented or neglected in discussions of the hallmarks of aging, Weissig synthesizes evidence showing that mitochondrial dysfunction is central to all twelve hallmarks of aging, including telomere attrition, genomic instability, epigenetic alterations, loss of proteostasis, cellular senescence, stem cell exhaustion, chronic inflammation, and more.
Marvin Edeas, Founder of WMS, states that Volkmar Weissig seeks to reposition mitochondrial dysfunction as the primary, “mother” hallmark of aging, arguing it underlies and drives all other mechanisms of aging. He calls for a paradigm shift in therapeutic strategy, moving beyond targeting isolated hallmarks toward preserving mitochondrial integrity and enhancing adaptive plasticity. It further emphasizes the need for anticipatory monitoring and systemic approaches to prevent cascading dysfunction across biological and societal levels.
We are also pleased to remind you that the World Mitochondria Society (WMS), in collaboration with the International Society of Microbiota (ISM), is organizing the Targeting Longevity World Congress 2026, taking place on April 8–9 in Berlin, Germany.
Biomolecular Condensates Found to Safeguard Mitochondria During Aging
- Details
- Published on 15 September 2025
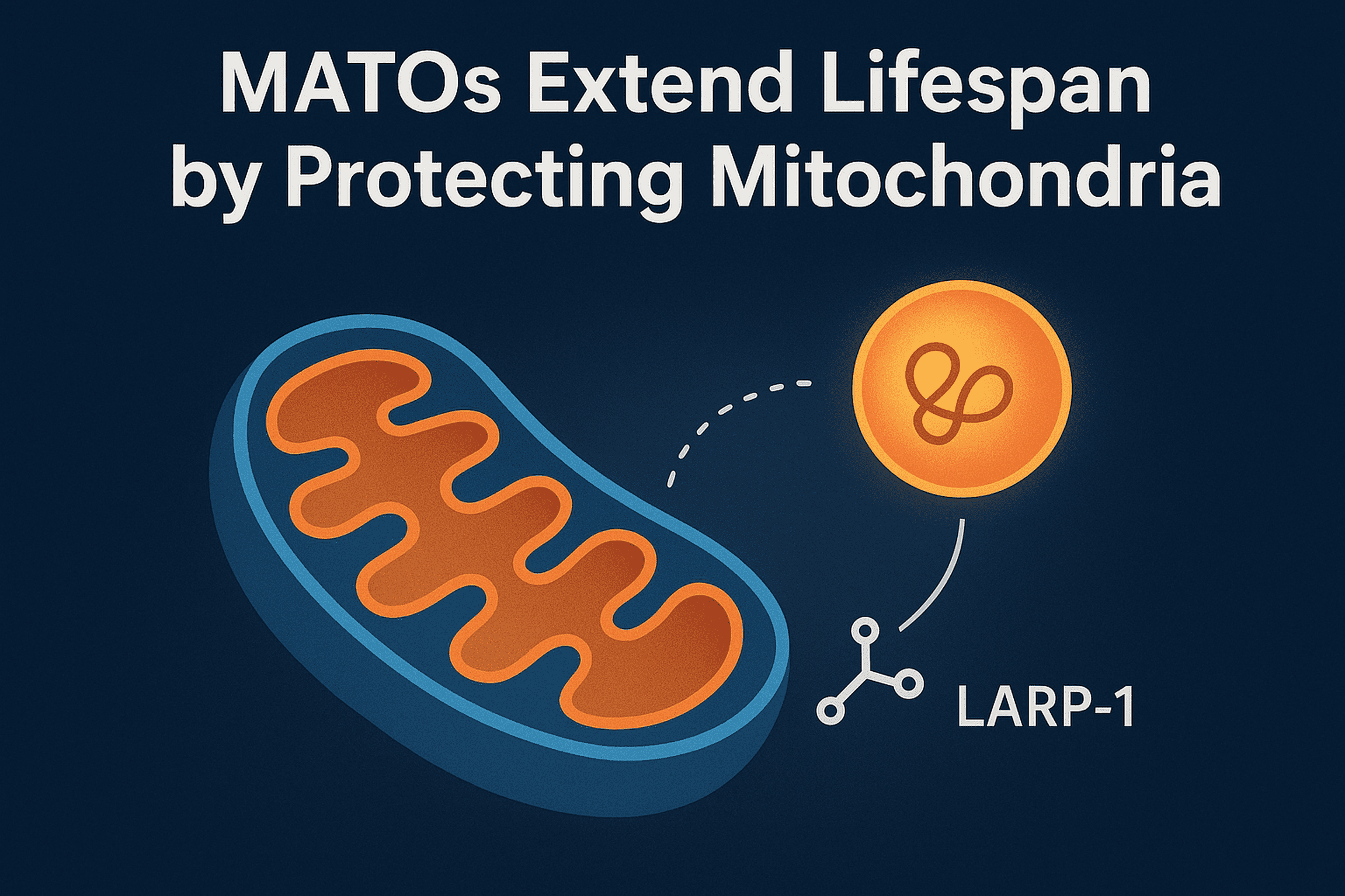
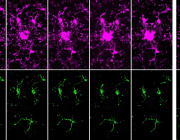
A new study in Nature Aging led by Prof. Chonglin Yang and colleagues has revealed a previously unknown mechanism that helps cells protect their powerhouses—mitochondria—during stress and aging.
The team discovered Mitochondria-Associated Translation Organelles (MATOs), a novel class of membraneless condensates formed by liquid–liquid phase separation (LLPS). These structures assemble directly on the mitochondrial surface, where they act as local protein factories.
At the center of this process is the RNA-binding protein LARP-1, which brings together ribosomes, mRNAs, and other RNA-binding proteins to produce crucial components for mitochondrial energy production and structure, such as IMMT-1 (a MICOS complex subunit for cristae organization) and ATP-2 (a key subunit of ATP synthase).
Using C. elegans as a model, the researchers showed that:
- Loss of LARP-1 or MATOs disrupts cristae organization, reduces ATP output, and shortens lifespan.
- By contrast, keeping MATOs anchored to mitochondria during stress or aging preserves mitochondrial function and significantly extends lifespan.
Mitochondrial decline is a hallmark of aging and contributes to neurodegeneration and metabolic disease. This discovery shows that biomolecular condensates directly regulate mitochondrial health, suggesting that stabilizing MATOs could become a new therapeutic strategy to combat age-related dysfunction.