Exploiting Mitochondrial Metabolic Vulnerabilities in Glioblastoma: Therapeutic Potential of Mubritinib
- Details
- Published on 25 February 2025


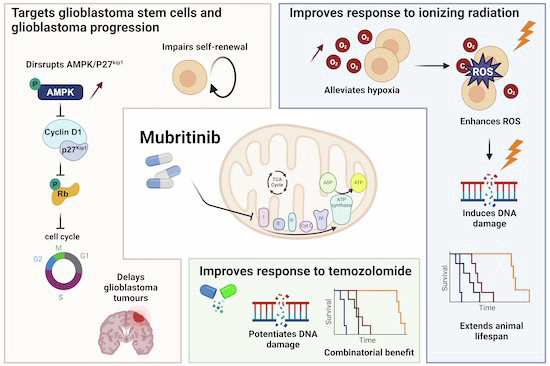
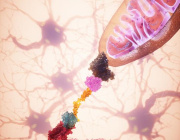
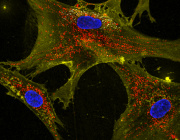
Glioblastoma remains one of the most treatment-resistant and lethal cancers, driven in part by a subset of self-renewing brain tumour stem cells (BTSCs) that contribute to therapy resistance and relapse. In a recent study, Ahmad Sharanek and his team from the University of Bordeaux and INSERM investigated the therapeutic potential of mubritinib in targeting BTSCs and glioblastoma progression by disrupting mitochondrial function.
The study demonstrated that mubritinib effectively impairs BTSC stemness and growth by targeting complex I of the electron transport chain. This disruption impairs self-renewal and proliferation, leading to a significant impact on glioblastoma progression. Gene expression profiling and Western blot analysis further revealed that mubritinib affects the AMPK/p27Kip1 pathway, resulting in cell-cycle impairment.
Importantly, pharmacokinetic assays confirmed that mubritinib crosses the blood-brain barrier, making it a viable candidate for glioblastoma therapy. Using preclinical patient-derived and syngeneic models, researchers observed that mubritinib delays tumour progression and extends survival. When combined with radiotherapy or chemotherapy, mubritinib provided an additional survival advantage.
A crucial finding was that mubritinib alleviates tumour hypoxia, enhancing reactive oxygen species (ROS) generation, DNA damage, and apoptosis when combined with radiotherapy. Toxicological and behavioural studies confirmed that mubritinib is well tolerated and spares normal cells, further supporting its potential as a therapeutic candidate.
This study highlights the promising therapeutic role of mubritinib in glioblastoma treatment by targeting mitochondrial respiration. By impairing mitochondrial function, mubritinib suppresses glioblastoma stem cells and tumour growth, while combination therapy with current standard treatments provides further therapeutic benefits. Given its well-tolerated profile and ability to penetrate the blood-brain barrier, mubritinib warrants further clinical exploration for glioblastoma therapy.
Image Credits: Burban et al. EMBO Molecular Medicine (2025)
Mitochondria: The Key to Stem Cell Longevity and Anti-Aging Strategies
- Details
- Published on 12 February 2025
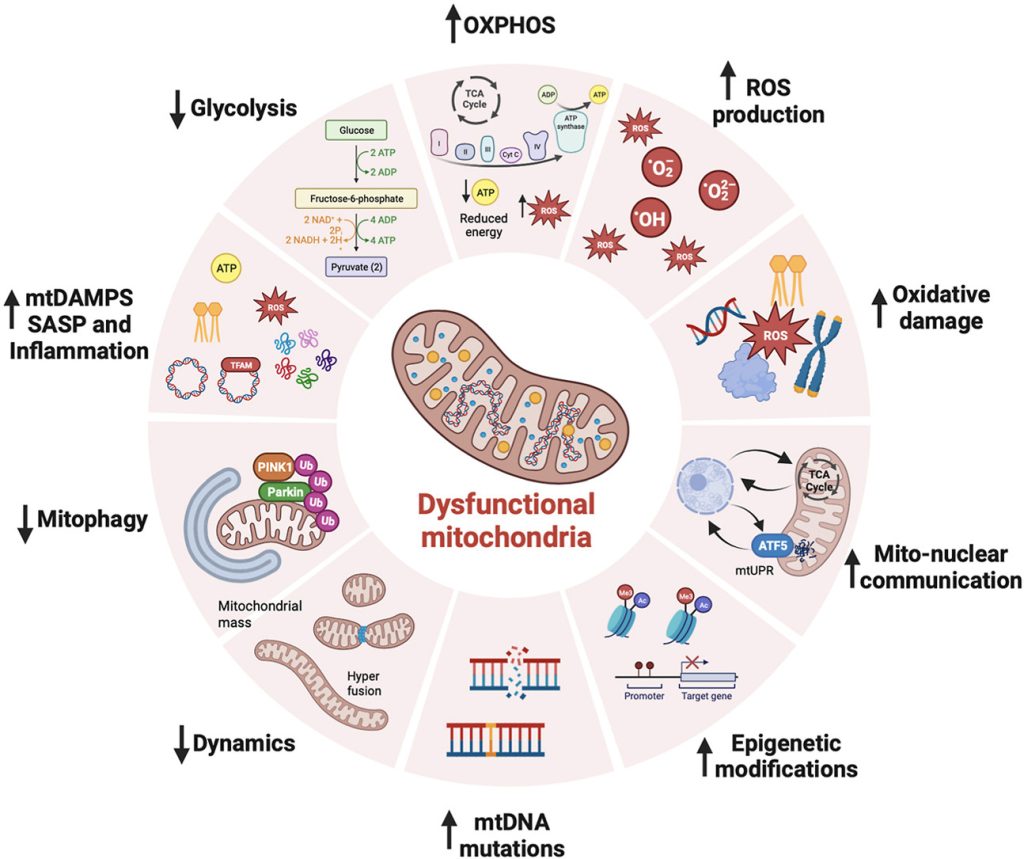
Mitochondria are at the heart of stem cell aging, influencing energy metabolism, oxidative stress, and cellular fate. As mitochondrial dysfunction accumulates with age, stem cells lose their regenerative potential, leading to tissue degeneration and age-related diseases. Cellular senescence is driven by increased ROS production, epigenetic modifications, and impaired mitophagy.
This review highlights innovative therapeutic strategies to counteract mitochondrial dysfunction, including antioxidants (MitoQ, nicotinamide riboside), mitochondrial biogenesis enhancers (AICAR, rapamycin), and mitophagy activators (Urolithin A, melatonin). Mitochondrial transfer from stem cells also emerges as a promising approach to restore function in aged tissues.
Targeting mitochondrial health represents a cutting-edge strategy to delay stem cell senescence and unlock new possibilities for regenerative medicine and longevity.
We remind you that we have a workshop dedicated to mitochondria transplantation during Targeting Mitochondria 2025 in Berlin. This hot topic will also be discussed during the 2nd World Congress on Targeting Longevity, where leading experts will explore the latest breakthroughs in mitochondrial research and anti-aging therapies.
For more details, read the full article here.

Mitochondria as a Reservoir of NAD⁺: Regulating Cellular Energy and Metabolic Stability
- Details
- Published on 10 February 2025
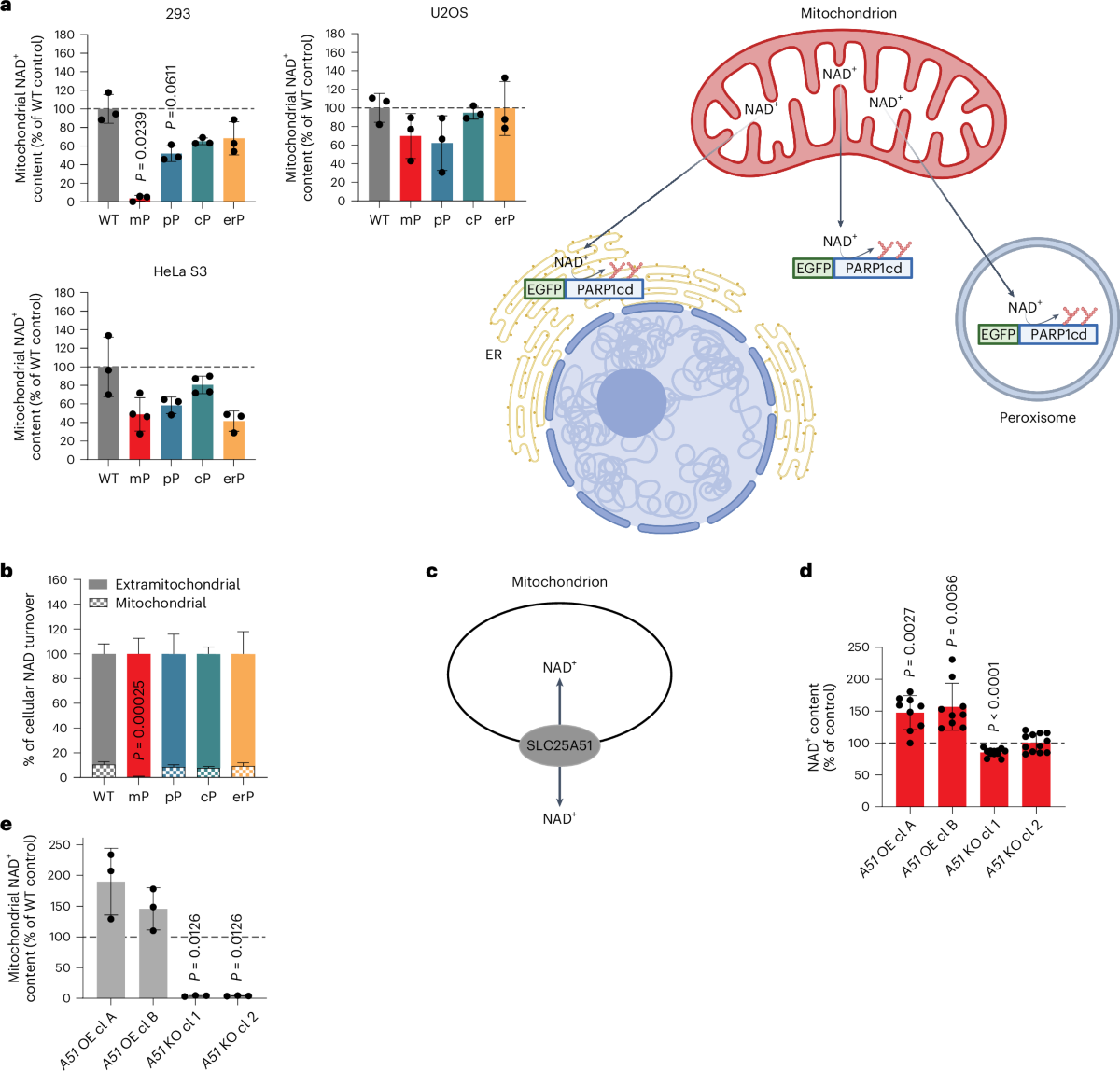
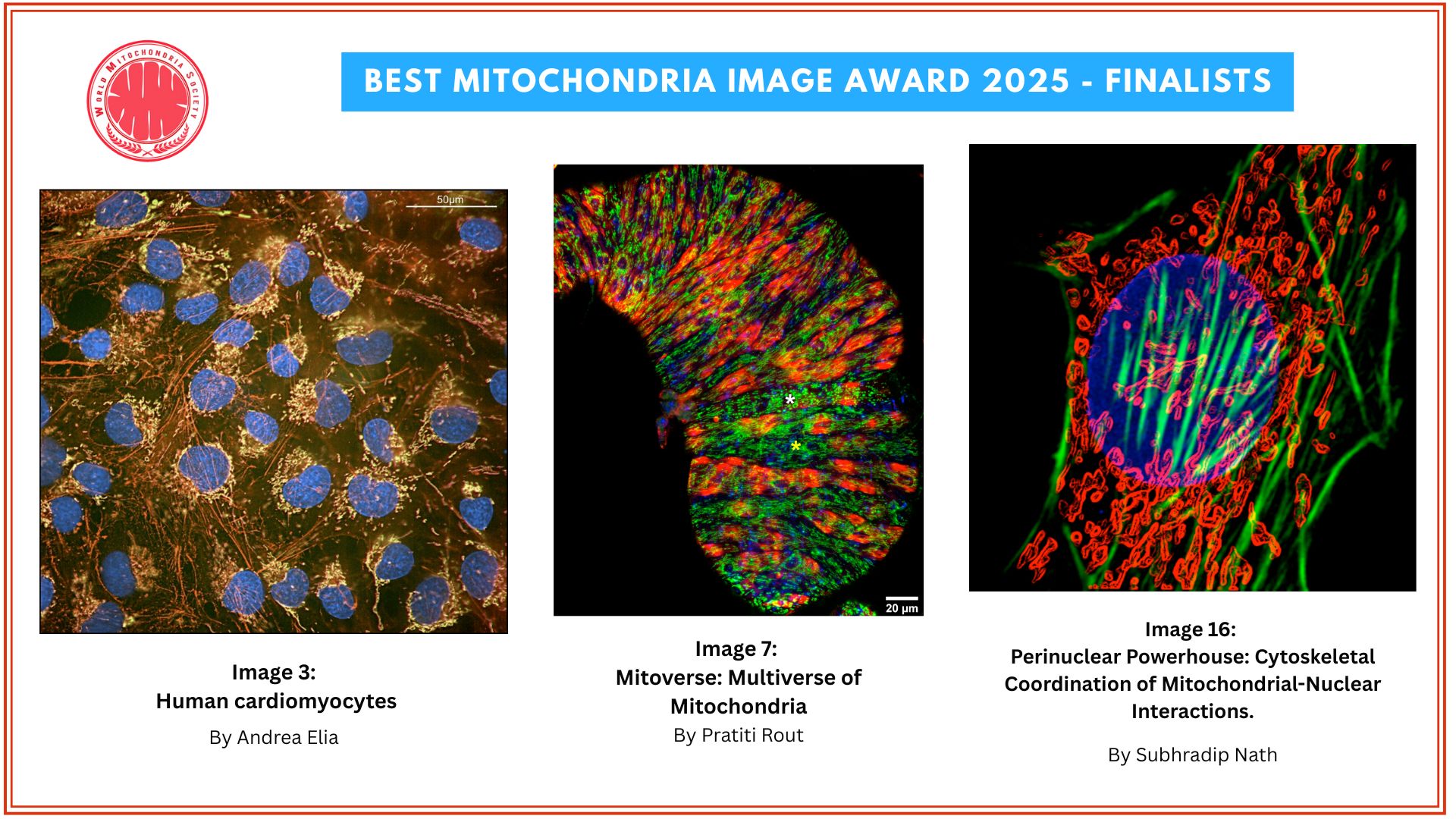
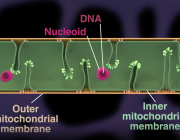
The mitochondrial NAD+ pool is regulated by SLC25A51 and is diminished in PARP1cd cell lines, irrespective of PARP1cd location
A paper published in Nature Metabolism by researchers from the University of Bergen (Norway) explored how cells manage their energy currency, specifically focusing on a molecule called NAD⁺ (nicotinamide adenine dinucleotide). NAD⁺ is essential for various cellular processes, including energy production and DNA repair.
Interconnected NAD⁺ Pools
The study reveals that different compartments within a cell, such as the nucleus, cytoplasm, and mitochondria, share and balance their NAD⁺ levels. This interconnectedness ensures that if one area experiences a decrease in NAD⁺, others can compensate to maintain overall cellular function.
Mitochondrial Role as a Buffer
Mitochondria, known as the cell’s powerhouses, play a crucial role in regulating NAD⁺ levels. They act as a buffer, adjusting their NAD⁺ content to stabilize the molecule’s availability throughout the cell, especially when there’s increased consumption elsewhere.
Implications for Cellular Health
Understanding this balancing act is vital because disruptions in NAD⁺ levels are linked to aging and various diseases. Insights from this research could inform strategies to maintain or restore NAD⁺ balance, potentially leading to therapeutic approaches for age-related conditions and metabolic disorders.
In summary, the study highlights the dynamic and interconnected nature of NAD⁺ distribution within cells, emphasizing the mitochondria’s pivotal role in maintaining cellular energy balance and overall health.
© Image: Høyland, L.E., VanLinden, M.R., Niere, M. et al. Subcellular NAD+ pools are interconnected and buffered by mitochondrial NAD+. Nat Metab 6, 2319–2337 (2024).
How Brain Cell Powerhouses Shape Our Memories: New Research Breakthrough
- Details
- Published on 10 February 2025
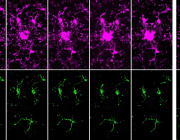
Scientists at Virginia Tech have made a fascinating discovery about how our brain cells manage energy during memory formation. Their research, published in Scientific Reports, reveals a crucial link between cellular powerhouses (mitochondria) and our ability to form memories, particularly those involving social interactions.
The Discovery
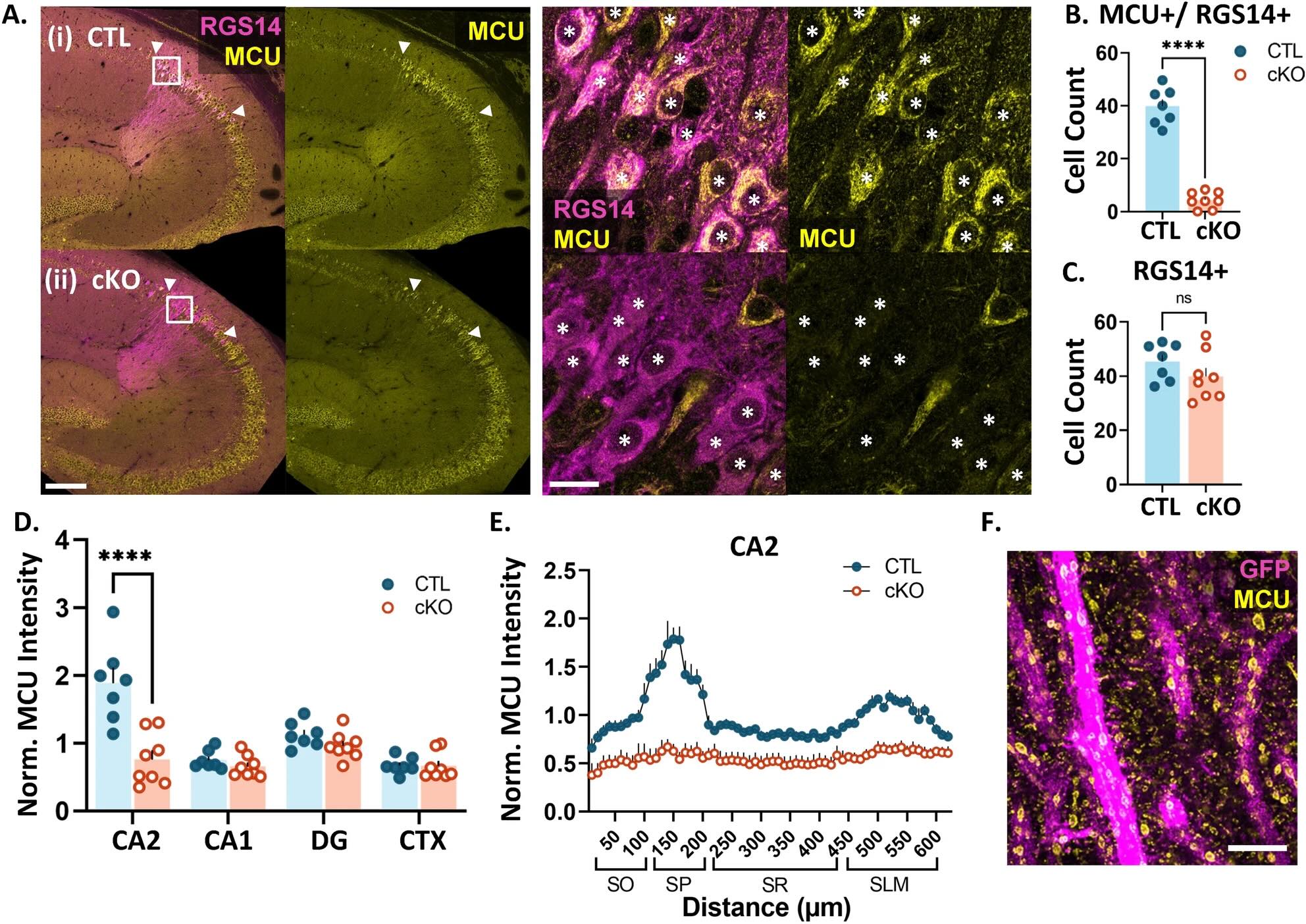
At the heart of this research is a tiny but mighty protein called MCU (mitochondrial calcium uniporter), which acts like a gatekeeper controlling calcium flow into mitochondria. The research team found this protein is especially abundant in the CA2 region of the hippocampus – our brain's social memory center.
What They Found
When researchers removed the MCU protein from brain cells, they observed several significant changes:
- Brain cells lost their ability to strengthen connections with each other
- Mitochondria became smaller and fragmented
- Memory-forming structures (dendritic spines) decreased in size
These changes essentially disrupted the brain's ability to form new memories effectively, highlighting just how important proper energy management is for brain function.
Why It Matters
This research is particularly exciting because it could help us better understand and potentially treat various memory-related conditions, including:
- Alzheimer's disease
- Autism spectrum disorders
- Schizophrenia
- Depression
- Bipolar disorder
Looking Forward
"Understanding how brain cells manage their energy needs during memory formation opens up new possibilities for treating memory disorders," explains Dr. Shannon Farris, the study's senior author. The team suggests that maintaining healthy mitochondrial function could be key to preserving memory and social behavior.
This groundbreaking research provides a new direction for developing treatments that target cellular energy systems, potentially offering hope to millions affected by memory-related disorders.
@ Image - Article: https://www.nature.com/articles/s41598-025-85958-4
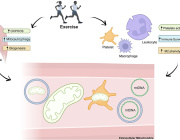
Tiny Messengers, Big Impact: Exosomes as Gatekeepers of Mitochondrial Function
- Details
- Published on 05 February 2025
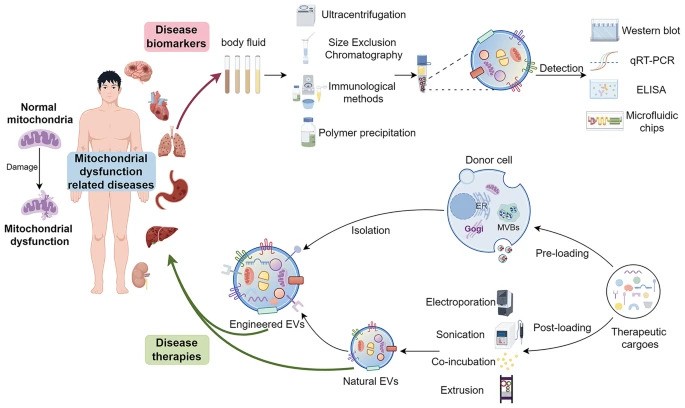
The article titled “Extracellular vesicles: opening up a new perspective for the diagnosis and treatment of mitochondrial dysfunction” was published in the Journal of Nanobiotechnology on August 14, 2024.
Key Points:
- Mitochondrial Dysfunction: Mitochondria are essential for energy production in eukaryotic cells. Disruptions such as oxidative stress, calcium imbalances, and mitochondrial DNA abnormalities can lead to mitochondrial dysfunction, contributing to various diseases.
- Extracellular Vesicles (EVs): EVs are cell-derived nanovesicles that facilitate intercellular communication by transporting proteins, lipids, and nucleic acids. They are categorized based on size into small EVs (sEVs, <200 nm) and large EVs (lEVs, >200 nm).
- EVs and Mitochondrial Components: Certain EV subtypes are enriched with mitochondrial components, including intact mitochondria. These EVs can transfer mitochondrial elements to recipient cells, influencing mitochondrial function.
- Modulation of Mitochondrial Function: EVs can affect mitochondrial homeostasis, apoptosis, and reactive oxygen species (ROS) generation in target cells by delivering specific substances.
- Therapeutic Potential: Research is exploring the artificial modification of EVs as delivery vehicles for therapeutic agents targeting mitochondria, offering new avenues for treating conditions associated with mitochondrial dysfunction.
Perspective:
This review highlights the emerging role of EVs in modulating mitochondrial function, presenting innovative strategies for diagnosing and treating diseases linked to mitochondrial dysfunction. By leveraging the natural communication pathways of EVs, there is potential to develop targeted therapies that restore mitochondrial health, offering hope for conditions such as neurodegenerative diseases, cardiovascular disorders, and cancer. Future research should focus on understanding the mechanisms governing EV-mediated mitochondrial modulation and developing safe, effective EV-based therapeutic approaches.
Learn more in the full article.
© Image: Li, J., Wang, T., Hou, X. et al. J Nanobiotechnol22, 487 (2024).
More Articles...
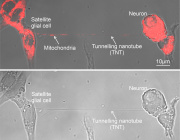
- Microglia Transfer Healthy Mitochondria to Rescue Neurons from Neurodegeneration
- The future of cancer immunotherapy will come through mitochondria: Cancer cells hijack T cells
- Targeting Mitophagy in Neurodegenerative Diseases: A Promising Therapeutic Avenue
- Ketone Bodies: A New Approach to Brain Function and Neurodegenerative Diseases