Mitochondria Detect Stress Earlier Than Previously Thought
- Details
- Published on 27 May 2026


-
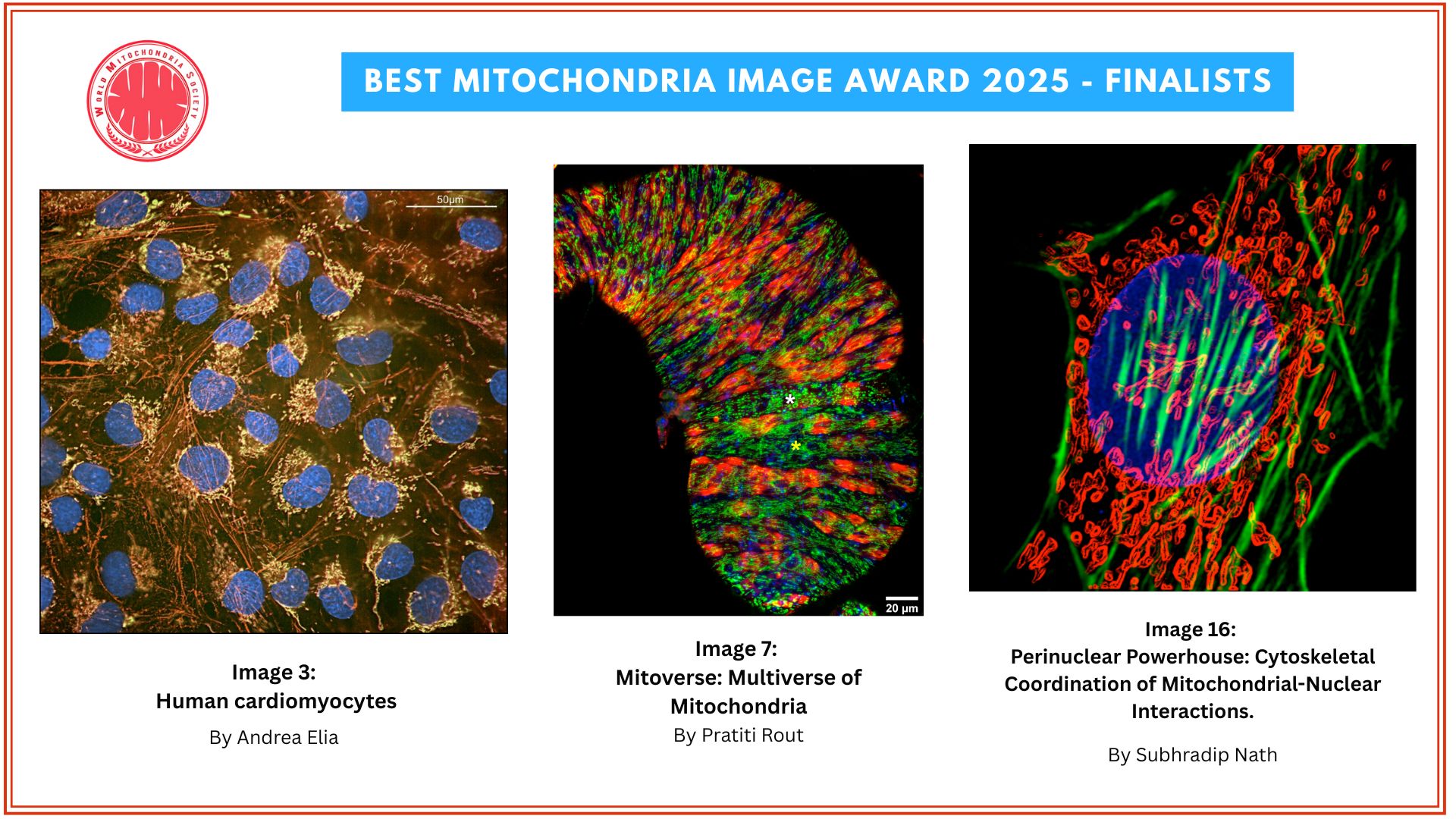
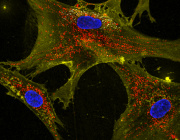
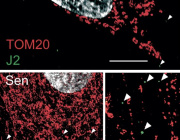
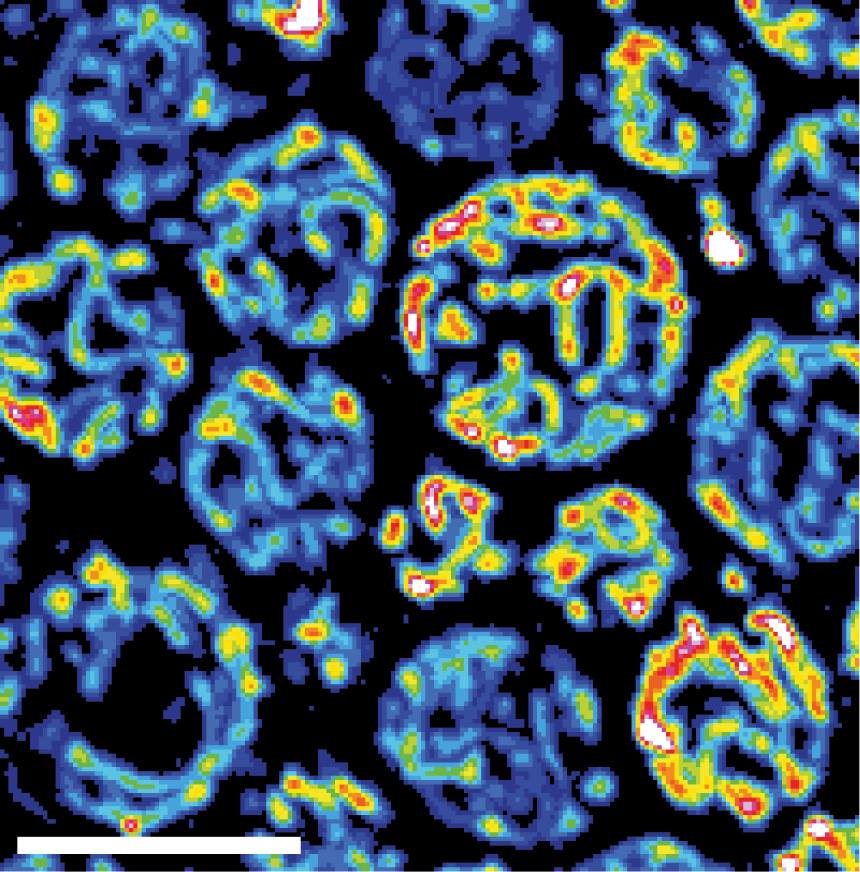
The mitochondrial network is shown in yeast cells. The reddish coloring indicates the fitness of the mitochondria. N. Photograph: Vögtle/ZMBH
“How Do Mitochondria Detect Danger Before Cellular Dysfunction Becomes Visible?”
Researchers from the University of Freiburg and Heidelberg University have identified a new mitochondrial early-warning system against oxidative stress.
The study was led by Prof. Dr Chris Meisinger, University of Freiburg, and Prof. Dr F.-Nora Vögtle, Heidelberg University. The first authors are Asli Aras Taskin and Sahana Shankar.
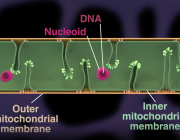
The team demonstrates that mitochondria can sense mild oxidative stress before visible cellular damage occurs. Under low-stress conditions, two mitochondrial protein-processing enzymes lose activity, leading to the formation of protein aggregates inside mitochondria. These aggregates function as internal alarm signals that activate the mitochondrial unfolded protein response (UPRmt).
The findings suggest that mitochondria are not passive victims of cellular stress, but active surveillance systems capable of initiating protective adaptation at very early stages.
Impact
The study reinforces a growing concept in mitochondrial medicine: pathology may begin not simply with mitochondrial failure, but with the disruption of mitochondrial sensing and adaptive signaling mechanisms.
A Conceptual Shift in Biology
This work marks a transition from a static view of mitochondria toward a systems-biology perspective, where mitochondria act as active coordinators of cellular decisions under stress.
The emerging question is no longer only:
“What happens when mitochondria are damaged?”
But increasingly:
“How do mitochondria detect danger before cellular dysfunction becomes visible?”
Statement from the World Mitochondria Society
According to Prof. Volkmar Weissig, President of the World Mitochondria Society, and Prof. Marvin Edeas, Chairman of the World Mitochondria Society:
“This study is not simply another mitochondrial dysfunction paper. It introduces the idea of intra-mitochondrial surveillance, which is highly aligned with the WMS conceptual evolution from: ‘What fuels mitochondria?’ to ‘What controls mitochondrial behavior?’”
Reference
Asli Aras Taskin, et Al, “Uncovering the Initial Response: Intra-mitochondrial Surveillance Activates the UPRmt.” Molecular Cell, 2026.
DOI: 10.1016/j.molcel.2026.05.002.
Leucine Rewrites Mitochondrial Biology
- Details
- Published on 21 May 2026
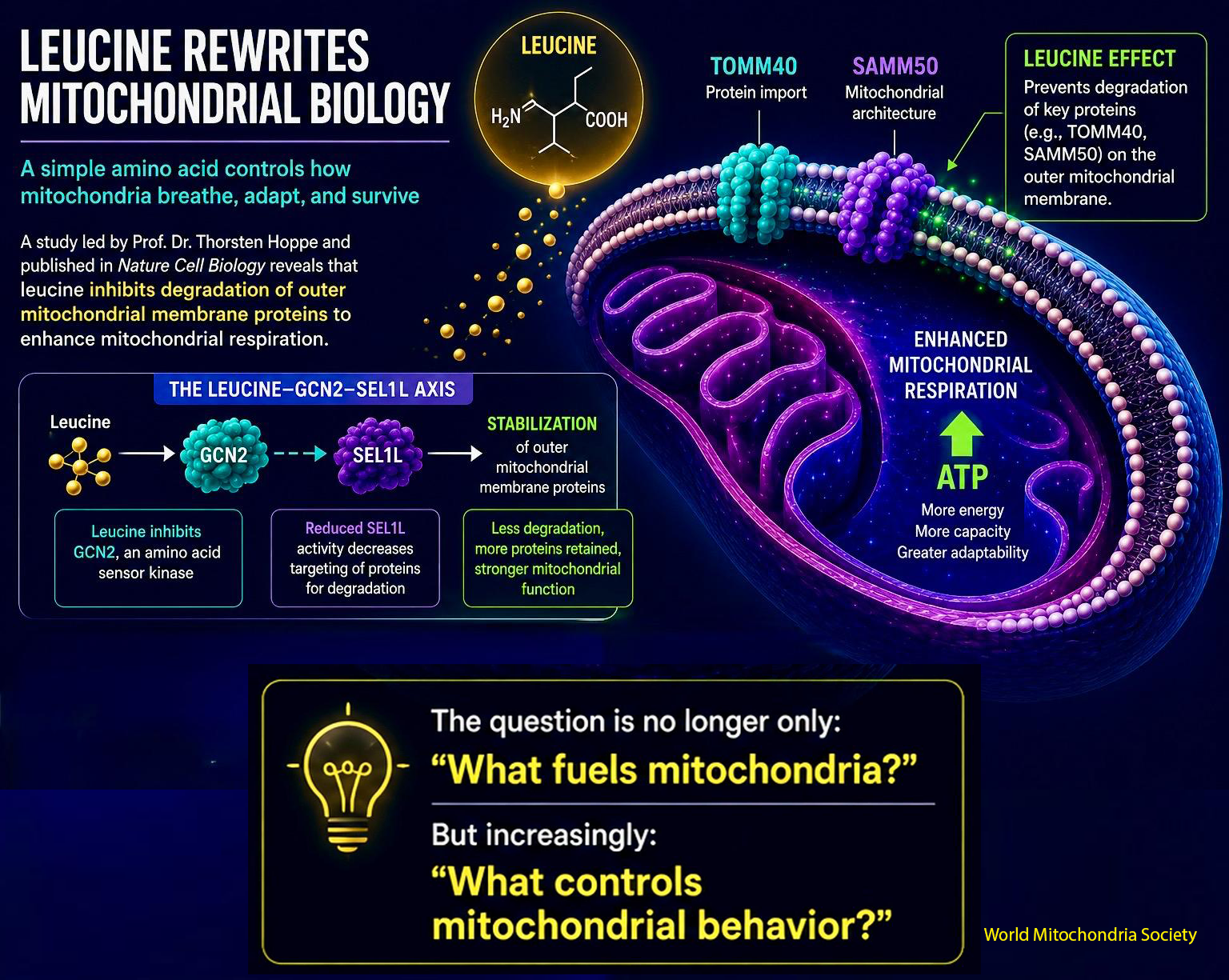
A simple amino acid may control how mitochondria breathe, adapt, and survive. From amino acid metabolism to mitochondrial decision-making.
A new study led by Professor Dr. Thorsten Hoppe from the Institute for Genetics and the CECAD Cluster of Excellence on Aging Research, published in Nature Cell Biology under the title “Leucine inhibits degradation of outer mitochondrial membrane proteins to adapt mitochondrial respiration”, reveals that leucine is doing far more than supporting muscle protein synthesis.
Researchers discovered that leucine directly remodels mitochondrial function by protecting key proteins on the outer mitochondrial membrane from degradation.
Not Just Fuel: A Nutrient Signal With Mitochondrial Power
Leucine emerges as a direct regulator of mitochondrial protein stability
The work identifies a previously unknown leucine–GCN2–SEL1L axis linking nutrient sensing to mitochondrial proteostasis and respiration. Instead of merely acting as a metabolic substrate, leucine appears to function as a regulator of mitochondrial adaptability.
The Hidden Switch on the Mitochondrial Surface
Stabilizing TOMM40 and SAMM50 to boost respiratory capacity
The team showed that leucine suppresses the degradation of outer mitochondrial membrane proteins, including TOMM40 and SAMM50, key components of mitochondrial protein import and architecture. This stabilization expands the mitochondrial proteome and enhances respiratory activity.
Beyond Nutrition: Toward Mitochondrial Resilience
Implications for metabolism, fertility, cancer, and aging biology
The findings extend beyond dietary biology. Defects in leucine metabolism or mitochondrial protein turnover were linked to fertility defects in C. elegans and enhanced survival of human cancer cells.
This reinforces a major emerging concept in mitochondrial medicine: nutrients are not only fuels. They can also program mitochondrial identity, adaptability, and resilience.
Strategic Perspective
The question is no longer only “What fuels mitochondria?” but “What controls mitochondrial behavior?”
For years, leucine has been viewed mainly through anabolic signaling and mTOR activation. This study shifts the discussion toward mitochondrial proteostasis, organelle remodeling, and adaptive bioenergetics.
Important scientific caution: this is a mechanistic study, not a clinical nutrition recommendation. As emphasized by Marvin Edeas and Volkmar Weissig, Chairmen of World Mitochondria Society and scientific Board, these findings should not be interpreted as evidence that leucine supplementation improves mitochondrial function in patients. Clinical relevance will require dedicated translational and human studies.
Metformin’s Mechanism Rewritten: Intestinal Mitochondria Take Center Stage
- Details
- Published on 12 May 2026
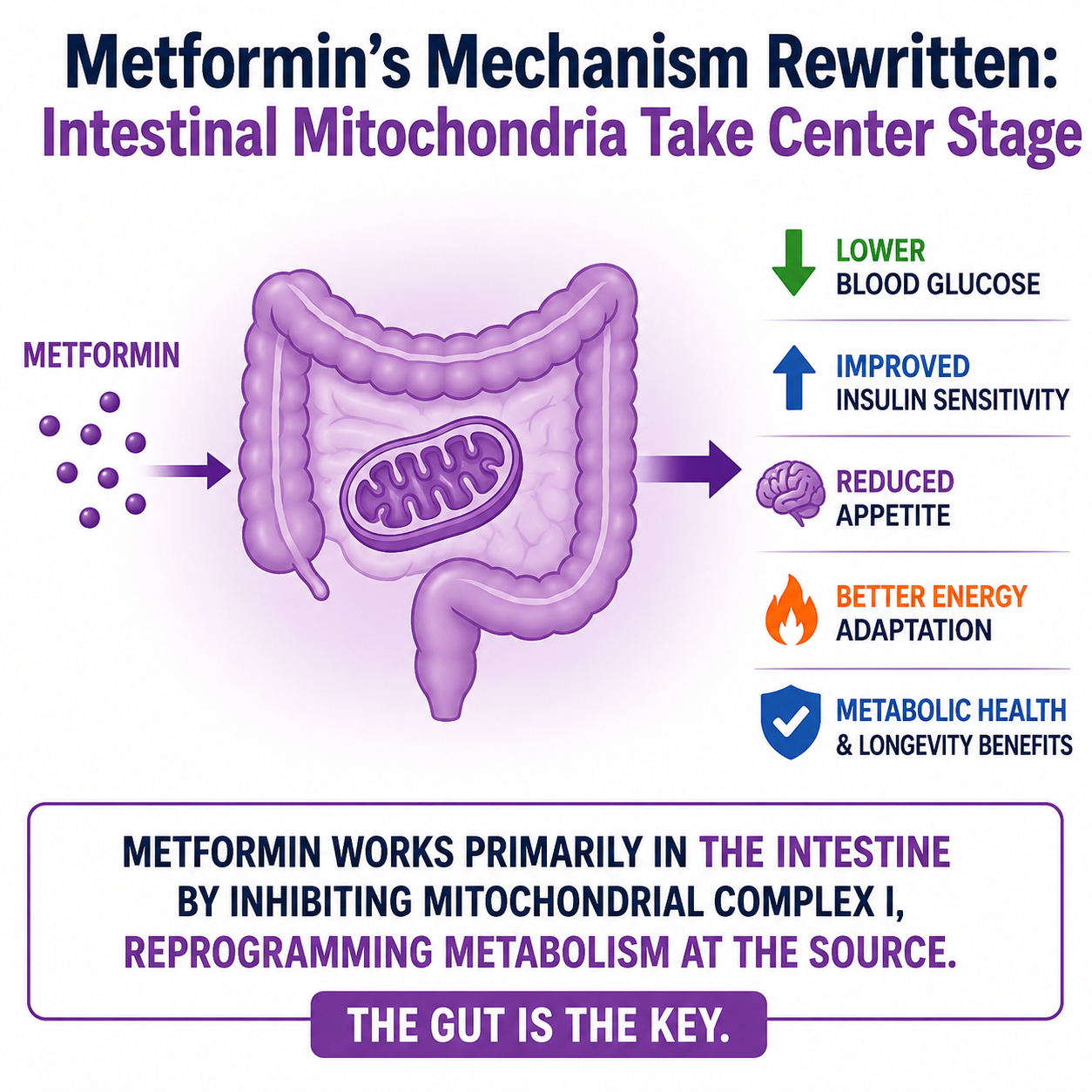 A new Nature Metabolism study challenges decades of assumptions about one of the world’s most prescribed drugs.
A new Nature Metabolism study challenges decades of assumptions about one of the world’s most prescribed drugs.
For decades, metformin has been described primarily as a liver-targeting diabetes drug that suppresses hepatic glucose production.
The study published by Navdeep S. Chandel, Zachary L. Sebo and colleagues from Northwestern University Feinberg School of Medicine, Chicago, USA proposes a major shift in this paradigm: metformin may exert its primary metabolic effects through the intestine by directly modulating mitochondrial function in gut epithelial cells.
The researchers demonstrate that metformin inhibits mitochondrial complex I within intestinal epithelial cells, triggering a metabolic reprogramming that increases intestinal glucose utilization and reshapes systemic glucose homeostasis.
This finding reframes metformin not simply as a glucose-lowering agent, but as a mitochondrial signaling modulator acting through the gut.
Beyond the Liver: A New View of Metformin
The study helps unify several long-standing observations associated with metformin treatment, including:
- Increased intestinal glucose uptake
- Altered lactate metabolism
- Gastrointestinal side effects
- Gut-mediated metabolic signaling
- Systemic adaptations linked to energy sensing
Rather than viewing these effects as secondary phenomena, the new work positions intestinal mitochondrial adaptation as a central therapeutic mechanism.
A Broader Shift in Mitochondrial Medicine.
The implications extend beyond diabetes.
The study reinforces an emerging concept in mitochondrial biology: therapeutic benefit may arise not from simply enhancing mitochondrial activity, but from precisely modulating mitochondrial stress and adaptation.
This aligns with growing interest in:
- Mitohormesis
- Adaptive bioenergetics
- Tissue-specific mitochondrial signaling
- Resilience-based therapeutic strategies
The findings may also influence future approaches in obesity, aging, metabolic disease, and longevity research, where the gut–mitochondria axis is increasingly recognized as a major regulator of systemic physiology.
A New Question for Metabolic Medicine
The work raises an important conceptual question for the field:
Should mitochondrial medicine focus on maximizing mitochondrial performance or on intelligently redirecting mitochondrial adaptation?
As mitochondrial biology moves from static models toward dynamic systems regulation, metformin may represent one of the earliest and most successful examples of therapeutic mitochondrial reprogramming.
Memory and Mitochondria: Are We Entering the Era of Programmable Brain Energy?
- Details
- Published on 17 May 2026

Memory and Mitochondria: Are We Entering the Era of Programmable Brain Energy?
Scientists remotely reactivated mitochondrial signaling and restored memory in dementia models, introducing a new paradigm for neurodegenerative therapy.
A new study published in Nature Neuroscience, led by Giovanni Marsicano and Luigi Bellocchio from NeuroCentre Magendie, INSERM and University of Bordeaux, together with Étienne Hébert-Chatelain from Université de Moncton, introduces a major conceptual shift in neurodegeneration research: instead of attempting only to protect damaged neurons, researchers directly reactivated mitochondrial signaling to restore brain function.
The work describes an engineered system called mitoDREADD-Gs, designed to selectively stimulate signaling pathways inside mitochondria.
Mitochondria generate ATP through oxidative phosphorylation. In neurons, this function is critical because synaptic transmission and memory formation require continuous energy. In Alzheimer’s disease and other neurodegenerative disorders, mitochondrial dysfunction, loss of membrane potential, impaired respiration, and metabolic failure are commonly observed.
To test whether restoring mitochondrial activity could directly improve cognition, the researchers engineered a synthetic GPCR-based receptor targeted to mitochondria. The system uses DREADD technology, meaning Designer Receptors Exclusively Activated by Designer Drugs, allowing mitochondrial signaling to be remotely activated using a synthetic ligand.
The receptor was fused to a mitochondrial targeting sequence and localized primarily to the outer mitochondrial membrane. Once activated, it stimulated mitochondrial Gs signaling, leading to increased cyclic AMP-associated pathways inside the organelle.
The consequences were significant:
- Increased mitochondrial membrane potential, Δψm
- Enhanced oxygen consumption rate, OCR
- Improved mitochondrial respiration
- Restoration of synaptic plasticity
- Recovery of memory performance in mouse models
Importantly, the intervention improved cognition not only in pharmacologically induced memory impairment, but also in mouse models of Alzheimer’s disease and frontotemporal dementia.
The study suggests that mitochondrial dysfunction may not simply be a downstream consequence of neurodegeneration, but an active driver of cognitive decline that can potentially be therapeutically reversed.
Scientifically, the work expands the emerging field of organelle-targeted signaling, where intracellular organelles are treated as programmable therapeutic platforms rather than passive structures.
This marks a departure from conventional mitochondrial medicine approaches centered mainly on antioxidants or damage prevention. Instead, the study proposes a new framework:
Mitochondria may become controllable bioenergetic hubs capable of dynamically modulating neuronal function.
This work raises a deeper question for neuroscience and mitochondrial medicine: if mitochondria can be remotely reactivated, are we still looking at dementia as a problem of irreversible neuronal loss, or as a failure of cellular energy control that may be corrected?
At the World Mitochondria Society, this strategic shift will be discussed through key questions:
Can cognitive decline be reversed by restoring mitochondrial bioenergetics before neurons are lost?
Are mitochondria becoming the next therapeutic control point in Alzheimer’s disease and dementia?
Although the work remains preclinical and was performed in mice, it opens new perspectives for mitochondrial neuromodulation, programmable bioenergetics, and future strategies aimed at restoring cognition through direct mitochondrial reactivation.
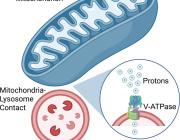
Mitochondria and Lysosomes Are Not Working Alone: Study Reveals Direct Proton Transfer Between Organelles
- Details
- Published on 11 May 2026
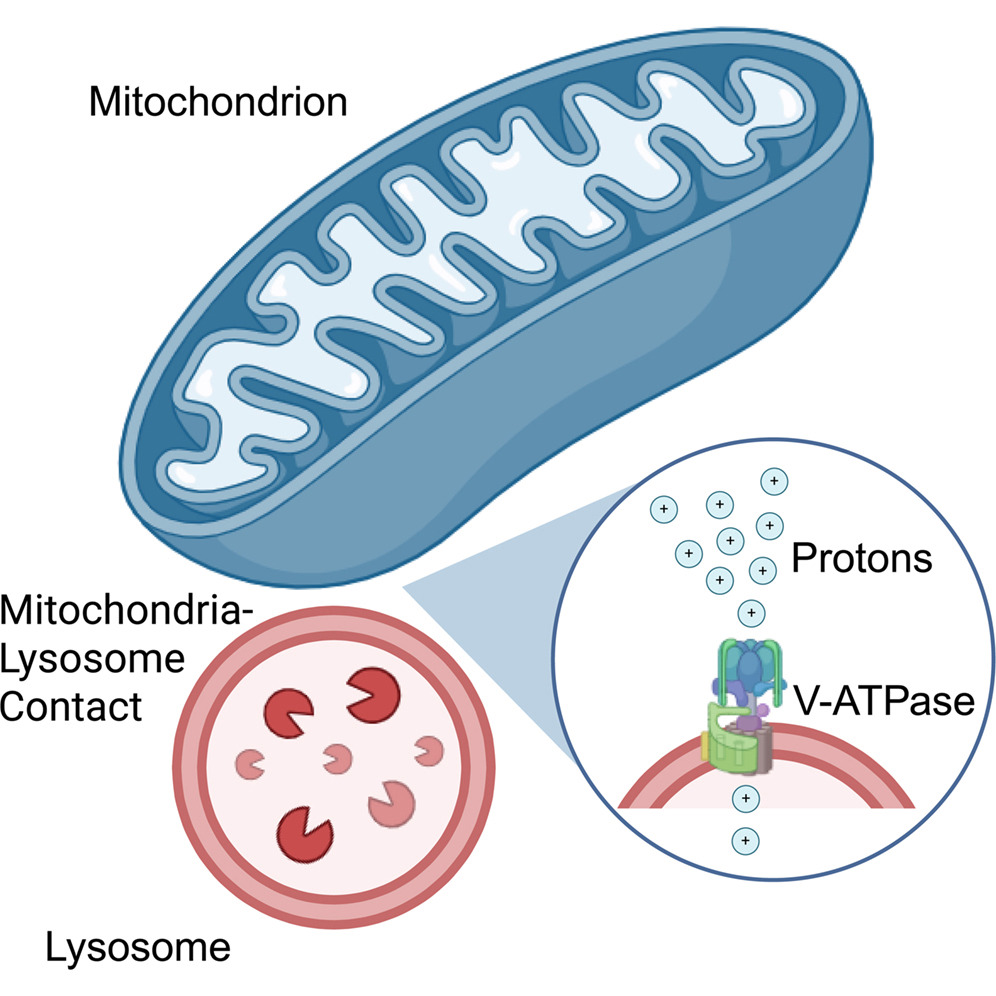
A new study by Jiajie Diao from the University of Cincinnati is reshaping how scientists understand communication between cellular organelles. Researchers discovered that mitochondria directly provide protons to lysosomes, revealing an unexpected metabolic cooperation inside animal cells.
For decades, biology textbooks described lysosomes as organelles that acidify themselves mainly by collecting protons from the surrounding cytosol. Acidification is essential because lysosomes function as the cell’s recycling and digestion system, breaking down damaged proteins, lipids, and cellular waste.
The new research, published in Cell Reports, suggests the process is far more coordinated.
Using a newly developed mitochondrial polarity probe, the team led by Jiajie Diao demonstrated that mitochondria act as major proton donors for lysosomal acidification.
This finding changes the traditional view of organelles as relatively independent cellular compartments. Instead, it reinforces the idea that cells operate through dynamic inter-organelle communication networks.
The implications could be substantial for aging and disease research.
Both mitochondrial dysfunction and lysosomal impairment are central features of neurodegenerative diseases, metabolic disorders, inflammation, and aging. The discovery that these two organelles are functionally coupled through proton transfer may open new therapeutic directions aimed at restoring cellular resilience rather than targeting isolated pathways.
The study also strengthens a growing scientific paradigm: mitochondria are not simply cellular “powerhouses.” They are signaling and coordination hubs that regulate broader cellular organization and adaptation.
Researchers now aim to understand how this proton-transfer mechanism is regulated and whether dysfunction of mitochondria–lysosome communication contributes directly to disease progression.
More Articles...
- Pancreatic Cancer’s Hidden Weakness: Damaged Mitochondria May Be Its Achilles’ Heel
- Can Mitochondria Create New Organelles? A Discovery Reopens a Major Question in Cell Biology
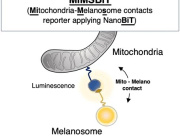
- Mitochondria Help Control Skin Pigmentation: A New Organelle Dialogue Revealed
- Red Blood Cells Lose Their Mitochondria: But Not Before Building the Machinery of Oxygen Transport