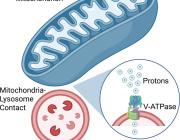
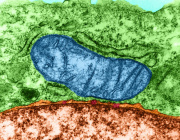
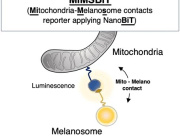
Parkinson's, cancer, type 2 diabetes share a key element that drives disease
- Details
- Published on 03 May 2021


When cells are stressed, chemical alarms go off, setting in motion a flurry of activity that protects the cell's most important players. During the rush, a protein called Parkin hurries to protect the mitochondria, the power stations that generate energy for the cell. Now Salk researchers have discovered a direct link between a master sensor of cell stress and Parkin itself. The same pathway is also tied to type 2 diabetes and cancer, which could open a new avenue for treating all three diseases.
"Our findings represent the earliest step in Parkin's alarm response that anyone's ever found by a long shot. All the other known biochemical events happen at one hour; we've now found something that happens within five minutes," says Professor Reuben Shaw, director of the NCI-designated Salk Cancer Center and senior author of the new work, detailed in Science Advances on April 7, 2021. "Decoding this major step in the way cells dispose of defective mitochondria has implications for a number of diseases."
Parkin's job is to clear away mitochondria that have been damaged by cellular stress so that new ones can take their place, a process called mitophagy. However, Parkin is mutated in familial Parkinson's disease, making the protein unable to clear away damaged mitochondria. While scientists have known for some time that Parkin somehow senses mitochondrial stress and initiates the process of mitophagy, no one understood exactly how Parkin was first sensing problems with the mitochondria -- Parkin somehow knew to migrate to the mitochondria after mitochondrial damage, but there was no known signal to Parkin until after it arrived there.
Shaw's lab, which is well known for their work in the fields of metabolism and cancer, spent years intensely researching how the cell regulates a more general process of cellular cleaning and recycling called autophagy. About ten years ago, they discovered that an enzyme called AMPK, which is highly sensitive to cellular stress of many kinds, including mitochondrial damage, controls autophagy by activating an enzyme called ULK1.
Following that discovery, Shaw and graduate student Portia Lombardo began searching for autophagy-related proteins directly activated by ULK1. They screened about 50 different proteins, expecting about 10 percent to fit. They were shocked when Parkin topped the list. Biochemical pathways are usually very convoluted, involving up to 50 participants, each activating the next. Finding that a process as important as mitophagy is initiated by only three participants -- first AMPK, then ULK1, then Parkin -- was so surprising that Shaw could scarcely believe it.
To confirm the findings were correct, the team used mass spectrometry to reveal precisely where ULK1 was attaching a phosphate group to Parkin. They found that it landed in a new region other researchers had recently found to be critical for Parkin activation but hadn't known why. A postdoctoral fellow in Shaw's lab, Chien-Min Hung, then did precise biochemical studies to prove each aspect of the timeline and delineated which proteins were doing what, and where. Shaw's research now begins to explain this key first step in Parkin activation, which Shaw hypothesizes may serve as a "heads-up" signal from AMPK down the chain of command through ULK1 to Parkin to go check out the mitochondria after a first wave of incoming damage, and, if necessary, trigger destruction of those mitochondria that are too gravely damaged to regain function.
The findings have wide-ranging implications. AMPK, the central sensor of the cell's metabolism, is itself activated by a tumor suppressor protein called LKB1 that is involved in a number of cancers, as established by Shaw in prior work, and it is activated by a type 2 diabetes drug called metformin. Meanwhile, numerous studies show that diabetes patients taking metformin exhibit lower risks of both cancer and aging comorbidities. Indeed, metformin is currently being pursued as one of the first ever "anti-aging" therapeutics in clinical trials.
"The big takeaway for me is that metabolism and changes in the health of your mitochondria are critical in cancer, they're critical in diabetes, and they're critical in neurodegenerative diseases," says Shaw, who holds the William R. Brody Chair. "Our finding says that a diabetes drug that activates AMPK, which we previously showed can suppress cancer, may also help restore function in patients with neurodegenerative disease. That's because the general mechanisms that underpin the health of the cells in our bodies are way more integrated than anyone could have ever imagined."
News Source: www.sciencedaily.com
Article: Chien-Min Hung, Portia S. Lombardo, Nazma Malik, Sonja N. Brun, Kristina Hellberg, Jeanine L. Van Nostrand, Daniel Garcia, Joshua Baumgart, Ken Diffenderfer, John M. Asara, Reuben J. Shaw. AMPK/ULK1-mediated phosphorylation of Parkin ACT domain mediates an early step in mitophagy. Science Advances, 2021; 7 (15): eabg4544 DOI: 10.1126/sciadv.abg4544
Bird blood is a heating system in winter
- Details
- Published on 03 May 2021

Credit: Dr. Andreas Nord, researcher in evolutionary ecology at Lund University, Sweden
Researchers at Lund University in Sweden have discovered that bird blood produces more heat in winter, when it is colder, than in autumn. The study is published in The FASEB Journal.
The secret lies in the energy factories of cells, the mitochondria. Mammals have no mitochondria in their red blood cells, but birds do, and according to the research team from Lund and Glasgow this means that the blood can function as a central heating system when it is cold.
"In winter, the mitochondria seem to prioritize producing more heat instead of more energy. The blood becomes a type of radiator that they can turn up when it gets colder," says Andreas Nord, researcher in evolutionary ecology at Lund University who led the study.
Until now, the common perception has been that birds keep warm by shivering with their large pectoral muscles and fluffing up their feathers. Less is known about other heat-regulating processes inside birds.
To investigate the function of mitochondria, the researchers examined great tits, coal tits and blue tits on two different occasions: early autumn and late winter. The researchers took blood samples from the birds and isolated the red blood cells. By using a so-called cell respirometer, a highly sensitive instrument that can measure how much oxygen the mitochondria consume, the researchers were able to calculate how much of the oxygen consumption was spent on producing energy and how much was spent on creating heat. Finally, they also measured the amount of mitochondria in each blood sample.
The results show that the blood samples taken in winter contained more mitochondria and that the mitochondria worked harder. However, the work was not to produce more energy, something the researchers had assumed since birds have a much higher metabolism in winter.
"We had no idea that the birds could regulate their blood as a heating system in this way, so we were surprised," says Andreas Nord.
The researchers will now investigate whether cold weather is the whole explanation for the birds' blood producing more heat in winter. Among other things, they will study whether the food that the birds eat in winter affects the mitochondria.
Dr. Andreas Nord will join the Targeting Mitochondria 2021 Congress and will present his study on Avian red blood cell mitochondria produce more heat in winter than in autumn.News source: www.sciencedaily.com
Article: Andreas Nord, Neil B. Metcalfe, Jennifer L. Page, Anna Huxtable, Dominic J. McCafferty, Neal J. Dawson. Avian red blood cell mitochondria produce more heat in winter than in autumn. The FASEB Journal, 2021; 35 (5) DOI: 10.1096/fj.202100107R
Scientists solve a 100-year-old mystery about cancer
- Details
- Published on 03 May 2021

The year 2021 marks the 100th anniversary of a fundamental discovery that's taught in every biochemistry textbook. In 1921, German physician Otto Warburg observed that cancer cells harvest energy from glucose sugar in a strangely inefficient manner: rather than "burn" it using oxygen, cancer cells do what yeast do -- they ferment it. This oxygen-independent process occurs quickly, but leaves much of the energy in glucose untapped.
Various hypotheses to explain the Warburg effect have been proposed over the years, including the idea that cancer cells have defective mitochondria -- their "energy factories" -- and therefore cannot perform the controlled burning of glucose. But none of these explanations has withstood the test of time. (Cancer cells' mitochondria work just fine, for example.)
Now a research team at the Sloan Kettering Institute led by immunologist Ming Li offers a new answer, based on a hefty set of genetic and biochemical experiments and published January 21 in the journal Science.
It comes down to a previously unappreciated link between Warburg metabolism and the activity of a powerhouse enzyme in the cell called PI3 kinase.
"PI3 kinase is a key signaling molecule that functions almost like a commander-in-chief of cell metabolism," Dr. Li says. "Most of the energy-costly cellular events in cells, including cell division, occur only when PI3 kinase gives the cue."
As cells shift to Warburg metabolism, the activity of PI3 kinase is increased, and in turn, the cells' commitment to divide is strengthened. It's a bit like giving the commander-in-chief a megaphone.
The findings revise the commonly accepted view among biochemists that sees metabolism as secondary to cell signaling. They also suggest that targeting metabolism could be an effective way to thwart cancer growth.
Challenging the Textbook View
Dr. Li and his team, including graduate student Ke Xu, studied Warburg metabolism in immune cells, which also rely on this seemingly inefficient form of metabolism. When immune cells are alerted to the presence of an infection, a certain type called T cells shift from the typical oxygen-burning form of metabolism to Warburg metabolism as they grow in number and ramp up infection-fighting machinery.
The key switch that controls this shift is an enzyme called lactate dehydrogenase A (LDHA), which is made in response to PI3 kinase signaling. As a result of this switch, glucose remains only partially broken down and the cell's energy currency, called ATP, is quickly generated in the cell's cytosol. (In contrast, when cells use oxygen to burn glucose, the partially broken down molecules travel to the mitochondria and are further broken down there to make ATP on a delay.)
Dr. Li and his team found that in mice, T cells lacking LDHA could not sustain their PI3 kinase activity, and as a result could not effectively fight infections. To Dr. Li and his team, this implied that this metabolic enzyme was controlling a cell's signaling activity.
"The field has worked under the assumption that metabolism is secondary to growth factor signaling," Dr. Li says. "In other words, growth factor signaling drives metabolism, and metabolism supports cell growth and proliferation. So the observation that a metabolic enzyme like LDHA could impact growth factor signaling through PI3 kinase really caught our attention."
Like other kinases, PI3 kinase relies on ATP to do its work. Since ATP is the net product of Warburg metabolism, a positive feedback loop is set up between Warburg metabolism and PI3 kinase activity, securing PI3 kinase's continued activity -- and therefore cell division.
As for why activated immune cells would preferentially resort to this form of metabolism, Dr. Li suspects it has to do with the cells' need to produce ATP quickly to ramp up their cell division and infection-fighting machinery. The positive feedback loop ensures that once this program is engaged, it will be sustained until the infection is eradicated.
The Cancer Connection
Though the team made their discoveries in immune cells, there are clear parallels to cancer.
"PI3 kinase is a very, very critical kinase in the context of cancer," Dr. Li says. "It's what sends the growth signal for cancer cells to divide, and is one of the most overly active signaling pathways in cancer."
As with immune cells, cancer cells may employ Warburg metabolism as a way to sustain the activity of this signaling pathway and therefore ensure their continued growth and division. The results raise the intriguing possibility that doctors could curb cancer growth by blocking the activity of LDHA -- the Warburg "switch."
News source: www.sciencedaily.com
Article: Ke Xu, Na Yin, Min Peng, Efstathios G. Stamatiades, Amy Shyu, Peng Li, Xian Zhang, Mytrang H. Do, Zhaoquan Wang, Kristelle J. Capistrano, Chun Chou, Andrew G. Levine, Alexander Y. Rudensky, Ming O. Li. Glycolysis fuels phosphoinositide 3-kinase signaling to bolster T cell immunity. Science, 2021; 371 (6527): 405 DOI: 10.1126/science.abb2683
Rapid blood test identifies COVID-19 patients at high risk of severe disease
- Details
- Published on 03 May 2021
One of the most vexing aspects of the COVID-19 pandemic is doctors' inability to predict which newly hospitalized patients will go on to develop severe disease, including complications that require the insertion of a breathing tube, kidney dialysis or other intensive care. Knowledge of a patient's age and underlying medical conditions can help predict such outcomes, but there are still surprises when younger, seemingly healthier patients suffer severe complications that can lead to death.
Now, scientists at Washington University School of Medicine in St. Louis have shown that a relatively simple and rapid blood test can predict -- within a day of a hospital admission -- which patients with COVID-19 are at highest risk of severe complications or death.
The study, published Jan. 14 in JCI Insight, involved nearly 100 patients newly admitted to the hospital with COVID-19.
The blood test measures levels of mitochondrial DNA, a unique type of DNA molecule that normally resides inside the energy factories of cells. Mitochondrial DNA spilling out of cells and into the bloodstream is a sign that a particular type of violent cell death is taking place in the body.
"Doctors need better tools to evaluate the status of COVID-19 patients as early as possible because many of the treatments -- such as monoclonal antibodies -- are in short supply, and we know that some patients will get better without intensive treatments," said co-senior author Andrew E. Gelman, PhD, the Jacqueline G. and William E. Maritz Endowed Chair in Immunology and Oncology in the Department of Surgery.
"There's so much we still don't understand about this disease," he added. "In particular, we need to understand why some patients, irrespective of their ages or underlying health in some cases, go into this hyperinflammatory death spiral. Our study suggests that tissue damage may be one cause of this spiral, since the mitochondrial DNA that is released is itself an inflammatory molecule."
The researchers said the test could serve as a way to predict disease severity as well as a tool to better design clinical trials, identifying patients who might, for example, benefit from specific investigational treatments. They also said they would like to evaluate whether the test could serve as a way to monitor the effectiveness of new therapies. Presumably, effective treatments would lower mitochondrial DNA levels.
"We will need larger trials to verify what we found in this study, but if we could determine in the first 24 hours of admission whether a patient is likely to need dialysis or intubation or medication to keep their blood pressure from dropping too low, that would change how we triage the patient, and it might change how we manage them much earlier in the disease course," said co-senior author Hrishikesh S. Kulkarni, MD, an assistant professor of medicine.
The researchers, including co-first authors Davide Scozzi, MD, PhD, a staff scientist, and Marlene Cano, PhD, a postdoctoral research scholar, evaluated 97 patients with COVID-19 at Barnes-Jewish Hospital, measuring their mitochondrial DNA levels on the first day of their hospital stays. They found that mitochondrial DNA levels were much higher in patients who eventually were admitted to the ICU, intubated or died. The researchers found this association held independently of a patient's age, sex and underlying health conditions.
On average, mitochondrial DNA levels were about tenfold higher in patients with COVID-19 who developed severe lung dysfunction or eventually died. Those with elevated levels were almost six times more likely to be intubated, three times more likely to be admitted to the ICU and almost twice as likely to die compared with those with lower levels.
Further, the test predicted outcomes as well as or better than existing markers of inflammation currently measured in patients hospitalized with COVID-19. Most other markers of inflammation measured in patients with COVID-19, including those still under investigation, are general markers of systemic inflammation, rather than inflammation specific to cell death, according to the researchers.
"Viruses can cause a type of tissue damage called necrosis that is a violent, inflammatory response to the infection," Gelman said. "The cell breaks open, releasing the contents, including mitochondrial DNA, which itself drives inflammation. In COVID-19 patients, there has been anecdotal evidence of this type of cell and tissue damage in the lung, heart and kidney. We think it's possible that measures of mitochondrial DNA in the blood may be an early sign of this type of cell death in vital organs."
The researchers also emphasized that the test is quick and straightforward to perform in most hospital settings because it uses the same machinery that processes the standard PCR test for COVID-19. The method they developed allows mitochondrial DNA levels to be quantified directly in the blood. Without requiring intermediate steps to extract the DNA from the blood, the technique returned results in less than an hour.
Before they can apply for approval from the Food and Drug Administration (FDA), the scientists will need to verify that the test is accurate in a larger multi-center trial. They have plans to expand the research to more sites.
The study utilized samples obtained from the School of Medicine's COVID-19 biorepository, which was developed by co-authors Jane O'Halloran, MD, PhD, an assistant professor of medicine; Charles Goss, PhD, an instructor in biostatistics; and Phillip Mudd, MD, PhD, an assistant professor of emergency medicine.
This work was supported by the Barnes Jewish Hospital Foundation; the Children's Discovery Institute; the National Institutes of Health (NIH), grant numbers, R01HL094601, P01AI116501 and K08HL148510; and the Washington University Institute of Clinical and Translational Sciences (ICTS) COVID-19 Research Program, which is funded by the National Center for Advancing Translational Sciences (NCATS) of the NIH, grant number UL1TR002345.
News source: www.sciencedaily.com
Article source: Davide Scozzi, Marlene Cano, Lina Ma, Dequan Zhou, Ji Hong Zhu, Jane A. O’Halloran, Charles W. Goss, Adriana M. Rauseo, Zhiyi Liu, Sanjaya Kumar Sahu, Valentina Peritore, Monica Rocco, Alberto Ricci, Rachele Amodeo, Laura Aimati, Mohsen Ibrahim, Ramsey R. Hachem, Daniel Kreisel, Philip A. Mudd, Hrishikesh S. Kulkarni, Andrew E. Gelman. Circulating mitochondrial DNA is an early indicator of severe illness and mortality from COVID-19. JCI Insight, 2021; DOI: 10.1172/jci.insight.143299
New possibilities to prevent sudden cardiac death
- Details
- Published on 03 May 2021

Nearly a half-million people a year die from sudden cardiac death (SCD) in the U.S. -- the result of malfunctions in the heart's electrical system.
A leading cause of SCD in young athletes is arrhythmogenic cardiomyopathy (ACM), a genetic disease in which healthy heart muscle is replaced over time by scar tissue (fibrosis) and fat.
Stephen Chelko, an assistant professor of biomedical sciences at the Florida State University College of Medicine, has developed a better understanding of the pathological characteristics behind the disease, as well as promising avenues for prevention. His findings are published in the current issue of Science Translational Medicine.
Individuals with ACM possess a mutation causing arrhythmias, which ordinarily are non-fatal if managed and treated properly. However, Chelko shows that exercise not only amplifies those arrhythmias, but causes extensive cell death. Their only option is to avoid taking part in what should be a healthy and worthwhile endeavor: exercise.
"There is some awful irony in that exercise, a known health benefit for the heart, leads to cell death in ACM subjects," Chelko said. "Now, we know that endurance exercise, in particular, leads to large-scale myocyte cell death due to mitochondrial dysfunction in those who suffer from this inherited heart disease."
Several thousand mitochondria are in nearly every cell in the body, processing oxygen and converting food into energy. Considered the powerhouse of all cells (they produce 90 percent of the energy our bodies need to function properly), they also play another important role as a protective antioxidant.
As mitochondria fail to function properly, and myocyte cells in the heart die, healthy muscles are replaced by scar tissue and fatty cells. Eventually, the heart's normal electrical signals are reduced to an erratic and disorganized firing of impulses from the lower chambers, leading to an inability to properly pump blood during heavy exercise. Without immediate medical treatment, death occurs within minutes.
Chelko's research gets to the heart of the process involved in mitochondrial dysfunction.
"Ultimately, mitochondria become overwhelmed and expel 'death signals' that are sent to the nucleus, initiating large-scale DNA fragmentation and cell death," Chelko said. "This novel study unravels a pathogenic role for exercise-induced, mitochondrial-mediated cell death in ACM hearts."
In addition to providing a better understanding of the process involved, Chelko discovered that cell death can be prevented by inhibiting two different mitochondrial proteins. One such approach utilizes a novel targeting peptide developed for Chelko's research by the National Research Council in Padova, Italy.
That discovery opens avenues for the development of new therapeutic options to prevent myocyte cell death, cardiac dysfunction and the pathological progression leading to deadly consequences for people living with ACM.
This research was funded by an American Heart Association Career Development Award.
News source: www.sciencedaily.com