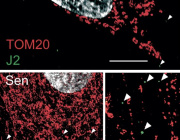
Know thy mitochondria: Autoimmunity to organelles and their DNA
- Details
- Published on 08 January 2020



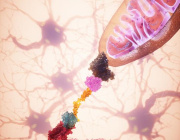
VDAC1 oligomerization forms a pore for mtDNA. Credit: Jeonghan Ki
The immune system uses its mitochondria to self-stimulate innate and adaptive responses to infection. Reactive oxygen species (ROS), immunogenic mitochondrial DNA (mtDNA), and even whole mitochondria are locally mobilized in a delicate balance to create hot spots of inflammatory action. When normal limiting feedback on these processes is compromised, destructive autoimmune reactions often arise.
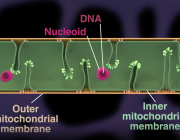
One common sign that all is not well in the immune system is the presence of antimitochondrial antibodies (AMAs) in the blood. In systemic lupus erthyematosis (SLE), for example, AMAs to several mitochondrial compartments can be found. Some AMAs target proteins normally found in the outer mitochondrial membrane while others seek out mtDNA. The question that naturally arises here is how the immune system sees mtDNA if it normally lives deep inside the mitochondrial matrix.
One answer was provided in a recent paper appearing in Science that shows released mtDNA can lead to lupus. In a nutshell, when mitochondria are stressed in various ways, mtDNA breaks into fragments and then binds to voltage-dependent anion channels (VDACs) in the outer membrane. This causes individual VDAC monomers to aggregate and form a meta-pore in their center through which the mtDNA can escape. Once in the cytoplasm, various nonspecific sensors including Toll receptors for single-stranded DNA, and the GAS-STING pathways for double-stranded DNA unleash a full-blown type I interferon (IFN) response.
Each VDAC monomer itself contains a highly-tuned channel that passes critical molecules of different sizes and charges in either direction according to the current prevailing membrane potential. Eliminating VDAC function entirely is incompatible with higher forms of eukaryotic life. Fortunately, the authors found that lupus-like immune activation can be eliminated by blocking just one form of the channel, the VDAC1 variety, with the oligomerization inhibitor VBIT-4.
The celebrity singer Seal suffers from discoid lupus erythematosis. It is a cutaneous form of lupus that is frequently related to SLE. As a general treatment strategy, interfering with mtDNA release alone might not fully eliminate all the AMAs someone like Seal might possess. However, for other forms of the disease, like lupus nephritis, such an approach might be more useful since all the AMAs found appear to be against double stranded mtDNA.
Other kinds of autoimmune disease, like those affecting the bile ducts of the liver, can similarly involve AMAs. Primary biliary cholangitis and primary sclerosing cholangitis are two such diseases that are characterized by different forms of autoantibodies. Sclerosing cholangitis, endured by the great NFL running back Walter Payton, is associated with antinuclear antibodies. On the other hand, biliary cholangitis sufferers have AMAs against the lipoate containing the E2 subunit of the pyruvate dehydrogenase complex. Additionally, they often also have antibodies to enzymes like sulfite oxidase and glycogen phosphorylase that are commonly associated to liver mitochondria.
At this point in time, it is not known exactly how and where these particular kinds of antibodies are made. The immunogenic E2 subunit normally floats inside the mitochondrial matrix along with the mtDNA and does not habitually escape through any channels. Presumably, errant pieces of degraded mitochondria from expiring cells could be contributing to antibody formation, as could whole mitochondria purposively ejected from cells.
One glaring problem in trying to understand the generation of AMAs that we have overlooked here so far is how the mtDNA gets past the inner membrane to reach the outer membrane VDAC. I asked Peggy Crow, the author of the accompanying opinion piece to the main article, to comment on research in this area. She notes that although the exact answer is not known, imaging studies have revealed another pore system that acts much in parallel to VDAC. These so-called "BAK/BAX macropores' permit the inner mitochondrial membrane to herniate into the cytosol, permeabilize, and ferry matrix components, including mtDNA.
We haven't yet said much about the reactive oxygen species component of mitochondrial inflammation mentioned above. There is another recent finding that ties together oligomerization of the VDAC1 channel and ROS. More specifically, the authors of the study found that a molecule known as lipoxstatin-1 protects cells against ROS damage by reducing VDA1 and restoring levels of the enzyme GPX4.
GPX4 is a unique selenium-utilizing form of glutathione peroxidase that specifically protects cytosolic membrane lipids from oxidative damage. When GPX4 is compromised, a unique form of apoptosis known as ferroptosis occurs throughout the cell. By blocking oligomerization of VDAC1, and not VDAC2 or 3, liproxstatin-1 was found to short circuit the ferroptosis pathway.
Importantly, liproxistatin also blocked the shrinkage of mitochondria, reduction and disruption of their cristae, and rupture of the outer mitochondrial membrane that keynotes ferroptosis. GPX4 deficiency is not an autoimmune disease but rather a disease of runaway ROS damage and ferroptosis within extremely circumscribed cell populations. It is extremely rare—so rare, in fact, that when one patient was recently diagnosed, he was the only person in the world to have this enigmatic disease.
News source: https://medicalxpress.com/
New Fluorescent Marker Allows Detailed Study of Mitochondria in Live Cells, Study Reports
- Details
- Published on 08 January 2020
MitoPB Yellow, a new fluorescent specific marker, allows scientists to study live mitochondria, possibly leading to a better understanding of mitochondrial diseases and to new treatments, researchers in Japan report.
The marker is much more stable than current markers, and allows mitochondria to be studied for extended periods without inducing toxicity to living cells.
The study, “A photostable fluorescent marker for the superresolution live imaging of the dynamic structure of the mitochondrial cristae,” was published in the journal PNAS.
Mitochondria, the small cellular structures that produce energy for our cells, are made of an inner and an outer membrane. The enzymes that produce ATP (the molecular “currency” of energy) are located on the inner membrane. This membrane is folded into structures known as cristae, which increase its surface area.
STED microscopy is a type of super-resolution microscopy that allows a detailed look into the interior of cells. For this technique, researchers use fluorescent dyes to see the structures inside cells. But current dyes are not optimal, as they stop fluorescing with time, a process called photobleaching. This prevents researchers from observing cells over extended periods, and damaged markers can cause cells to die.
Researchers at Nagoya University’s Institute of Transformative Bio-Molecules have developed a new marker molecule, called MitoPB Yellow, which has a much longer lifespan under the microscope. This marker is specific for the inner mitochondrial membrane, making the mitochondria visible for longer periods of time.
Furthermore, the new marker allows researchers to detect structural changes of the mitochondrial inner membrane in live cells. Before, these structural changes could only be seen in dead cells.
In this study, researchers compared the MitoPB Yellow to other mitochondrial fluorescent markers. MitoPB Yellow showed greater stability — even after 50 images had been taken using the microscopy, researchers saw no significant decrease in the fluorescence intensity. Moreover, the intensity of MitoPB Yellow was maintained above 70%, the highest rating among the different mitochondrial markers.
To demonstrate the usefulness of MitoPB Yellow for imaging live cells, researchers placed mitochondria under conditions that are known to induce structural changes, which became visible using MitoPB Yellow.
Changes in mitochondria in response to deprivation of nutrients were also observed in great detail (high-resolution).
In this stressful environment, the inner mitochondrial membranes fused together both within a single mitochondrion and between neighboring mitochondria. This process may increase energy production while protecting these cellular structures from degradation.
By allowing for a more detailed study of live mitochondria, MitoPB Yellow may aid in understanding, diagnosing and developing therapies for mitochondrial disease.
“In summary, MitoPB Yellow surpasses currently used fluorescent mitochondrial markers,” the researchers wrote.
News source: https://mitochondrialdiseasenews.com
New cause of cell aging discovered
- Details
- Published on 26 July 2019

New research from the USC Viterbi School of Engineering could be key to our understanding of how the aging process works. The findings potentially pave the way for better cancer treatments and revolutionary new drugs that could vastly improve human health in the twilight years.
The work, from Assistant Professor of Chemical Engineering and Materials Science Nick Graham and his team in collaboration with Scott Fraser, Provost Professor of Biological Sciences and Biomedical Engineering, and Pin Wang, Zohrab A. Kaprielian Fellow in Engineering, was recently published in the Journal of Biological Chemistry.
"To drink from the fountain of youth, you have to figure out where the fountain of youth is, and understand what the fountain of youth is doing," Graham said. "We're doing the opposite; we're trying to study the reasons cells age, so that we might be able to design treatments for better aging."
What causes cells to age?
To achieve this, lead author Alireza Delfarah, a graduate student in the Graham lab, focused on senescence, a natural process in which cells permanently stop creating new cells. This process is one of the key causes of age-related decline, manifesting in diseases such as arthritis, osteoporosis and heart disease.
"Senescent cells are effectively the opposite of stem cells, which have an unlimited potential for self-renewal or division," Delfarah said. "Senescent cells can never divide again. It's an irreversible state of cell cycle arrest."
The research team discovered that the aging, senescent cells stopped producing a class of chemicals called nucleotides, which are the building blocks of DNA. When they took young cells and forced them to stop producing nucleotides, they became senescent, or aged.
"This means that the production of nucleotides is essential to keep cells young," Delfarah said. "It also means that if we could prevent cells from losing nucleotide synthesis, the cells might age more slowly."
Graham's team examined young cells that were proliferating robustly and fed them molecules labeled with stable isotopes of carbon, in order to trace how the nutrients consumed by a cell were processed into different biochemical pathways.
Scott Fraser and his lab worked with the research team to develop 3D imagery of the results. The images unexpectedly revealed that senescent cells often have two nuclei, and that they do not synthesize DNA.
Before now, senescence has primarily been studied in cells known as fibroblasts, the most common cells that comprised the connective tissue in animals. Graham's team is instead focusing on how senescence occurs in epithelial cells, the cells that line the surfaces of the organs and structures in the body and the type of cells in which most cancers arise.
Graham said that senescence is most widely known as the body's protective barrier against cancer: When cells sustain damage that could be at risk of developing into cancer, they enter into senescence and stop proliferating so that the cancer does not develop and spread.
"Sometimes people talk about senescence as a double-edged sword, that it protects against cancer, and that's a good thing," Graham said. "But then it also promotes aging and diseases like diabetes, cardiac dysfunction or atherosclerosis and general tissue dysfunction," he said.
Graham said the goal was not to completely prevent senescence, because that might unleash cancer cells.
"But then on the other hand, we would like to find a way to remove senescent cells to promote healthy aging and better function," he said.
Graham said that the team's research has applications in the emerging field of senolytics, the development of drugs that may be able to eliminate aging cells. He said that human clinical trials are still in early stages, but studies with mice have shown that by eliminating senescent cells, mice age better, with a more productive life span.
"They can take a mouse that's aging and diminishing in function, treat it with senolytic drugs to eliminate the senescent cells, and the mouse is rejuvenated. If anything, it's these senolytic drugs that are the fountain of youth," Graham said.
He added that in order for successful senolytic drugs to be designed, it was important to identify what is unique about senescent cells, so that drugs won't affect the normal, non-senescent cells.
"That's where we're coming in -- studying senescent cell metabolism and trying to figure out how the senescent cells are unique, so that you could design targeted therapeutics around these metabolic pathways," Graham said.
News Source : www.sciencedaily.com
Journal Reference:
Alireza Delfarah, Sydney Parrish, Jason A. Junge, Jesse Yang, Frances Seo, Si Li, John Mac, Pin Wang, Scott E. Fraser, Nicholas A. Graham. Inhibition of nucleotide synthesis promotes replicative senescence of human mammary epithelial cells. Journal of Biological Chemistry, 2019; 294 (27): 10564 DOI: 10.1074/jbc.RA118.005806
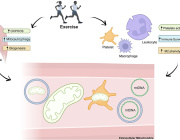
Autism Spectrum Disorders and Mitochondria
- Details
- Published on 06 September 2019

Mitochondria are the powerhouse that generate over 90% of the ATP produced in cells. In addition to its role in energy production, the mitochondrion also plays a major role in carbohydrate, fatty acid, amino acid and nucleotide metabolism, programmed cell death (apoptosis), generation of and protection against reactive oxygen species (ROS), immune response, regulation of intracellular calcium ion levels and even maintenance of gut microbiota. With its essential role in bio-energetic as well as non-energetic biological processes, it is not surprising that proper cellular, tissue and organ function is dependent upon proper mitochondrial function. Accordingly, mitochondrial dysfunction has been shown to be directly linked to a variety of medical disorders, particularly neuromuscular disorders and increasing evidence has linked mitochondrial dysfunction to neurodegenerative and neurodevelopmental disorders such as Alzheimer's Disease (AD), Parkinson's Disease (PD), Rett Syndrome (RS) and Autism Spectrum Disorders (ASD). Over the last 40 years there has been a dramatic increase in the diagnosis of ASD and, more recently, an increasing body of evidence indicates that mitochondrial dysfunction plays an important role in ASD development. In this review, the latest evidence linking mitochondrial dysfunction and abnormalities in mitochondrial DNA (mtDNA) to the pathogenesis of autism will be presented. This review will also summarize the results of several recent `approaches used for improving mitochondrial function that may lead to new therapeutic approaches to managing and/or treating ASD.
Abstract link: 10.1016/j.pnpbp.2018.12.015
During the Targeting Mitochondria 2019 Congress, we will talk about the relation between Autism Spectrum Disorders and Mitochondria.
For more info: https://targeting-mitochondria.com/
Broken mitochondria use 'eat me' proteins to summon their executioners
- Details
- Published on 26 July 2019
When mitochondria become damaged, they avoid causing further problems by signaling cellular proteins to degrade them. In a paper publishing April 11, 2019, in the journal Developmental Cell, scientists in Norway report that they have discovered how the cells trigger this process, which is called mitophagy. In cells with broken mitochondria, two proteins -- NIPSNAP 1 and NIPSNAP 2 -- accumulate on the mitochondrial surface, functioning as "eat me" signals, recruiting the cellular machinery that will destroy them.
NIPSNAP 1 and 2 are normally found inside healthy mitochondria, although their function inside the cell is unknown. "When a cell's respiration chain is disrupted, and the mitochondria are damaged, import of these proteins into the matrix and inner membrane space of the mitochondria is interrupted," says senior author Anne Simonsen, a professor at the Department of Molecular Medicine at the Institute of Basic Medical Sciences of the University of Oslo. "In that case, the import system does not function and they remain bound to the surface of the damaged mitochondria signaling for mitophagy."
In this study, the researchers studied human HeLa cells where both NIPSNAP1 and NIPSNAP 2 function were eliminated. "When we do that, these cells cannot clear the mitochondria after damage," says Simonsen. However, in cells with functional NIPSNAP proteins, when mitophagy was induced through the addition of a chemical disruptor, they observed that the NIPSNAP proteins act in concert with the PINK and PARKIN proteins, proteins already known to have a role in triggering autophagy and to have a role in Parkinson's Disease.
PARKIN labels cells with ubiquitin, a small protein that directs the cells towards degradation. "Ubiquitin is the classical signal to recruit autophagy," says co-author Terje Johansen, of the University of Tromsø -- The Arctic University of Norway. "What we saw is that in addition to ubiquitin, NIPSNAP proteins are required to recruit autophagy proteins; they are not targeted to the mitochondria unless these NIPSNAP proteins are found on the surface."
The team showed this finding has important physiological implications in vivo by investigating the NIPSNAP/PINK/PARKIN mechanism in a zebrafish animal model. They compared wild-type zebrafish and a fish line with reduced NIPSNAP1 protein abundance.
"We see that the mutant fish lacking adequate functional NIPSNAP1 are not able to move as the wild-type fish," says Simonsen. They have a Parkinsonian-like phenotype with reduced numbers of dopaminergic neurons. However, they could rescue this locomotion defect by adding L-dopa, the same compound used to treat human Parkinson's Disease, to the water.
Even more dramatically, animals entirely lacking NIPSNAP1 protein died within five days. "Clearly, clearance of mitochondria is important for the health of these dopaminergic neurons. That is particularly important since neurons generally cannot divide," says Johansen.
As evolutionarily conserved proteins, NIPSNAP proteins are found throughout the animal kingdom, including humans.
This work was partly funded by the Research Council of Norway, the Norwegian Cancer Society, and the Parkinson's Disease Foundation.
News Source : www.sciencedaily.com
Journal Reference:
Yakubu Princely Abudu, Serhiy Pankiv, Benan John Mathai, Alf Håkon Lystad, Christian Bindesbøll, Hanne Britt Brenne, Matthew Yoke Wui Ng, Bernd Thiede, Ai Yamamoto, Thaddaeus Mutugi Nthiga, Trond Lamark, Camila V. Esguerra, Terje Johansen, Anne Simonsen. NIPSNAP1 and NIPSNAP2 Act as 'Eat Me' Signals for Mitophagy. Developmental Cell, 2019; DOI: 10.1016/j.devcel.2019.03.013
More Articles...
- Researchers can finally modify plant mitochondrial DNA
- Caffeine from four cups of coffee protects the heart with the help of mitochondria
- Program of Targeting Mitochondria 2019 Congress/Speakers/Early Registration/May 21, 2019
- Marvin Edeas - A Pilot Study - Microbiota Quality and Mitochondrial Activity Link with Occurrence of Muscle Cramps ...