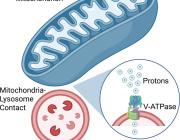
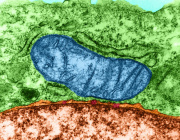
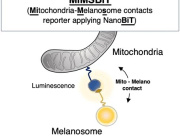
One in Five Brain Cancers Fueled by Overactive Mitochondria
- Details
- Published on 03 May 2021


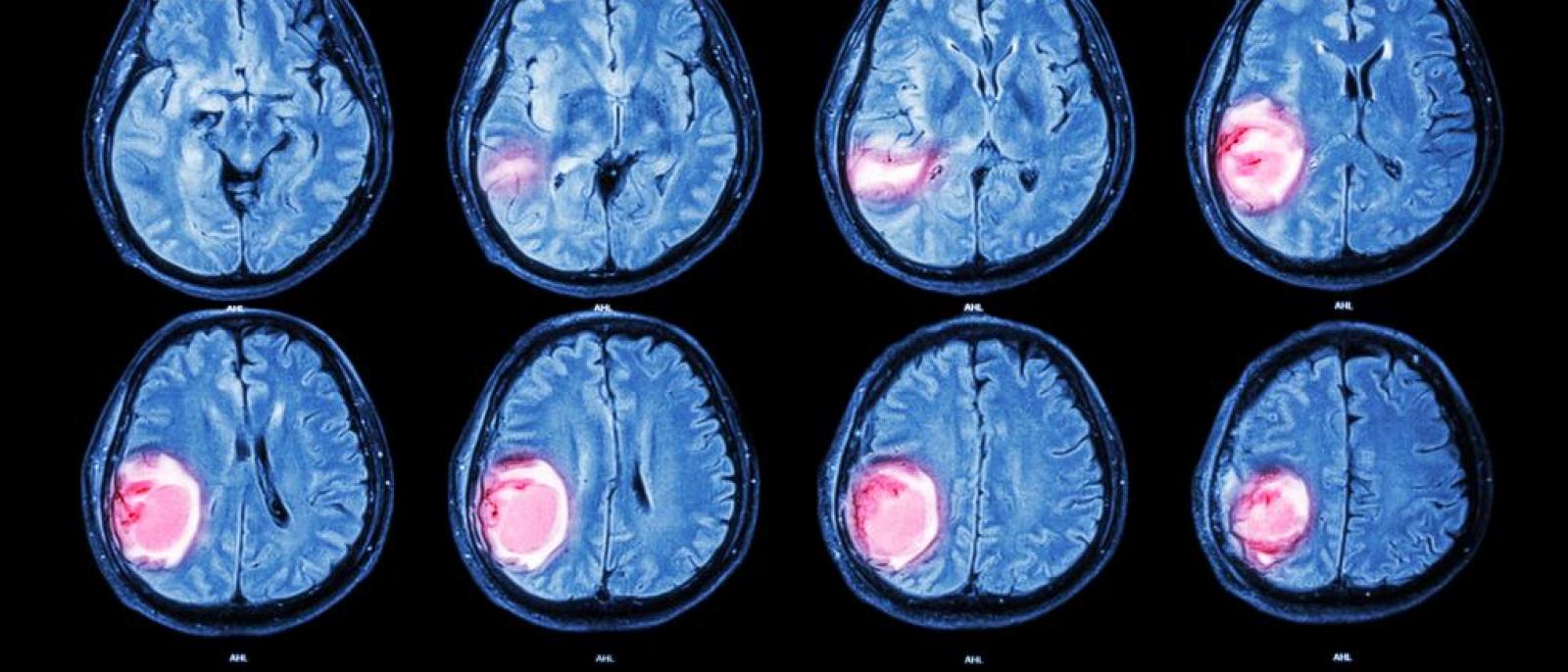
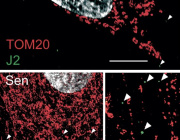
A new study(link is external and opens in a new window) has found that up to 20% of glioblastomas—an aggressive brain cancer—are fueled by overactive mitochondria and may be treatable with drugs currently in clinical trials.
Mitochondria are responsible for creating the energy that fuels all cells. Though they are usually less efficient at producing energy in cancer, tumor cells in this newly identified type of glioblastoma rely on the extra energy provided by overactive mitochondria to survive.
The study, by cancer scientists at Columbia University’s Vagelos College of Physicians and Surgeons and Herbert Irving Comprehensive Cancer Center, was published online Jan. 11 in Nature Cancer(link is external and opens in a new window).
The study also found that drugs that inhibit mitochondria—including a currently available drug and an experimental compound that are being tested in clinical trials—had a powerful anti-tumor effect on human brain cancer cells with overactive mitochondria. (Follow-up, unpublished work found that the same drugs are also active against mitochondrial tumors in glioblastomas growing in mice).
Such drugs are being tested in patients who have a rare gene fusion—previously discovered by the same researchers—that also sends mitochondria into overdrive.
“We can now expand these clinical trials to a much larger group of patients, because we can identify patients with mitochondria-driven tumors, regardless of the underlying genetics,” says Antonio Iavarone, MD, professor of neurology, who led the study with Anna Lasorella, MD, professor of pediatrics. Both are members of Columbia’s Institute for Cancer Genetics(link is external and opens in a new window).
Read more here: https://www.cuimc.columbia.edu/news/one-five-brain-cancers-fueled-overactive-mitochondria
Mitochondria found to be protected by ketogenesis
- Details
- Published on 03 May 2021

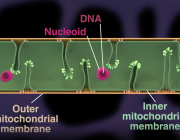
Ketone bodies are generally an alternative energy source during starvation, but in newborns, ketogenesis is active regardless of nutritional status. In a recent study from Kumamoto University (Japan), researchers analyzed the effects of ketogenesis in mice and found that it has a protective effect on cells by maintaining the function of mitochondria. They expect that this effect can be used in future therapies for protecting mitochondria and organs.
Ketones, along with glucose and fatty acids, are metabolites used as energy sources. In particular, ketones are known to be an alternative energy source during periods of fasting or starvation. However, ketogenesis is known to be active in the neonatal period regardless of the number of calories consumed during nursing, and role it plays in newborns is not well understood.
To search for answers, researchers generated ketogenesis-deficient mice that lacked the gene for HMG-CoA Synthase 2 (HMGCS2), an important enzyme ketogenesis. Their analysis showed that, in the absence of ketone bodies, the mice developed a severely fatty liver during the neonatal period.
Focusing on the mitochondria, they showed that enzymatic reactions in the mitochondria, mainly the Krebs cycle, were impaired. Nutrients are converted to acetyl CoA during the Krebs cycle, which is then converted to citric acid and seven other acids to produce energy. In the search for the cause of the dysfunction, researchers confirmed that the accumulation of the substrate acetyl CoA (due to insufficient ketogenesis) impairs the functions of proteins in the mitochondria by adding excessive acetylation.
"During a rapid increase in fatty acid intake with postnatal nursing, active ketogenesis under normal conditions has a protective effect by preventing excessive acetylation of mitochondrial proteins and maintaining mitochondrial function," said study leader Dr. Yuichiro Arima. "We believe that this result will be used in therapeutic applications for mitochondrial and organ protection in the future."
News source: www.sciencedaily.com
Article source: Yuichiro Arima, Yoshiko Nakagawa, Toru Takeo, Toshifumi Ishida, Toshihiro Yamada, Shinjiro Hino, Mitsuyoshi Nakao, Sanshiro Hanada, Terumasa Umemoto, Toshio Suda, Tetsushi Sakuma, Takashi Yamamoto, Takehisa Watanabe, Katsuya Nagaoka, Yasuhito Tanaka, Yumiko K. Kawamura, Kazuo Tonami, Hiroki Kurihara, Yoshifumi Sato, Kazuya Yamagata, Taishi Nakamura, Satoshi Araki, Eiichiro Yamamoto, Yasuhiro Izumiya, Kenji Sakamoto, Koichi Kaikita, Kenichi Matsushita, Koichi Nishiyama, Naomi Nakagata, Kenichi Tsujita. Murine neonatal ketogenesis preserves mitochondrial energetics by preventing protein hyperacetylation. Nature Metabolism, 2021; 3 (2): 196 DOI: 10.1038/s42255-021-00342-6
Dieting suppresses 'cellular engines', weight loss surgery gives boost to mitochondria
- Details
- Published on 03 May 2021

Mitochondria are important cellular power plants whose diminished activity has been previously demonstrated to be associated with obesity by a group of researchers at the University of Helsinki. Now, in a new international study coordinated by the University of Helsinki, the researchers have determined that the method of weight loss affects the metabolic pathways of mitochondria in fat tissue, also known as adipose tissue.
The study was recently published in the Journal of Clinical Endocrinology and Metabolism.
The researchers combined two datasets on calorie restriction diets and two datasets on weight loss surgery, or bariatric surgery, from Europe, monitoring dieters' weight loss as well as metabolism. A biopsy was taken from the study subjects' adipose tissue both at the beginning and the end of their weight reduction.
Ordinary dieting based on calorie restrictions put the mitochondria in the adipose tissue out of tune, further reducing the expression of related genes. In the case of similar weight loss resulting from bariatric surgery, the function of mitochondrial genes was improved and the activity level of mitochondrial metabolic pathways was higher.
The analyses conducted in the study were set in proportion to weight loss so that the results did not depend on greater weight loss in patients who had undergone surgery.
Why does lost weight come back? Impaired mitochondrial function is a potential cause.
Weight loss brings improvements to many metabolic changes associated with obesity, including disorders of glucose and lipid metabolism. Such beneficial effects were also observed in the new study, both in those who followed a regular diet and in those who underwent bariatric surgery.
"This is why it was astonishing to see that the activity of mitochondrial metabolic pathways in adipose tissue was entirely opposite in the two different groups," says researcher Birgitta van der Kolk from the University of Helsinki's Obesity Research Unit.
"Our observations indicate that impaired mitochondrial activity after losing weight by dieting may be the cause of adipose tissue rapidly building up again after weight loss. At the same time, bariatric surgery patients are better protected against regaining weight, which makes us suspect that a recovery of activity by mitochondria in the adipose tissue may be a factor underlying this phenomenon," says Professor Kirsi Pietiläinen, who led the study.
The study utilised a technique known as transcriptomics analysis, which makes it possible to read the genome as a whole.
"By combining these broad-based techniques, biocomputing and extensive European datasets, we observed entirely unexpected links between dieting and the mitochondria of adipose tissue. In the future, it is important to investigate the relevance of these mechanisms to the functioning of such tissue and weight regain," Pietiläinen adds.
News Source: www.sciencedaily.com/
Article source: Birgitta W van der Kolk, Maheswary Muniandy, Dorota Kaminska, Marcus Alvarez, Arthur Ko, Zong Miao, Armand Valsesia, Dominique Langin, Maija Vaittinen, Mirva Pääkkönen, Riikka Jokinen, Sanna Kaye, Sini Heinonen, Kirsi A Virtanen, Daniel P Andersson, Ville Männistö, Wim H Saris, Arne Astrup, Mikael Rydén, Ellen E Blaak, Päivi Pajukanta, Jussi Pihlajamäki, Kirsi H Pietiläinen. Differential Mitochondrial Gene Expression in Adipose Tissue Following Weight Loss Induced by Diet or Bariatric Surgery. The Journal of Clinical Endocrinology & Metabolism, 2021; DOI: 10.1210/clinem/dgab072
New data sheds light on genesis of our body's powerhouses
- Details
- Published on 03 May 2021

Scientists uncover for the first time how the body's energy makers are made using Cryo-Electron Microscopy (cryo-EM) at eBIC within Diamond which is based in Oxfordshire.
A new paper published in Science today (19 February 2021) by an international team of researchers reports an insight into the molecular mechanism of membrane-tethered protein synthesis in mitochondria. This is a fundamental new understanding of how the human mitoribosome functions and could explain how it is affected by mutations and deregulation that lead to disorders such as deafness and diseases including cancer development.
Mitochondria are intracellular organelles which serve as tiny but potent powerhouses in our body. They use oxygen which we inhale and derivatives from food we eat to produce more than 90% of our energy, and therefore effectively support our life. Mitochondria are particularly important in high-energy demanding organs such as heart, liver, muscles and brain. For example, almost 40% of each heart muscle cell is made up of mitochondria.
The bulk of energy production in mitochondria takes place in naturally evolved nano-factories incorporated in specialised membranes. These nano-factories consist of proteins cooperatively transporting ions and electrons to generate the chemical energy currency of our bodies which have to be constantly maintained, replaced and duplicated during cell division. To address this, mitochondria have their own protein making machine called the mitoribosome. The first fundamental understanding of how the mitoribosome looks was achieved in 2014.
"7 years ago, our work on the mitoribosome from yeast was termed the Resolution Revolution. The current study represents an additional advance on the original breakthrough. Not only does it reveal how the human mitoribosome is designed at an unprecedented level of detail, but it also explains the molecular mechanism that drives the process of bioenergetics to fuel life," says lead author, Alexey Amunts, Head of the program for Biology of Molecular Interactions, at SciLifeLab in Sweden.
The term Resolution Revolution was coined at Science magazine in relation to the first successful structure determination of the mitoribosome. This represented a methodological innovation in applying cryo-EM to understand molecular structures. However, this first glimpse into the architecture revealed only a partial picture of a static model. Yet the mitoribosome is a flexible molecular machine that requires the motion of its parts relative to each other in order to work. Therefore, in the current study, the team used the high throughput cryo-EM data acquisition at the electron Bio-Imaging Centre (eBIC) at Diamond to obtain 30 times more data allowing the team to describe conformational changes during the process of protein synthesis and association with the membrane adaptor. eBIC has been a strategic investment from the Wellcome Trust, UKRI's BBSRC and MRC. Being embedded at Diamond, eBIC benefits from amongst other things the well-established user support in place.
"Our study exposed the dynamic molecular mechanism that explains how the mitoribosome actually works to form the cellular powerhouse and reveals that the mitoribosome is much more flexible and active than previously thought. The discovery of intrinsic conformational changes represents a gating mechanism of the mitoribosome without similarity in bacterial and cytosolic systems. Together, the data offer a molecular insight into how proteins are synthesized in human mitochondria," adds Alexey Amunts.
Yuriy Chaban, Principal Electron Microscopy Scientist at eBIC, Diamond comments; "At Diamond, we are pushing the boundaries of what can be measured in the physical and life sciences and this latest development is tribute to the team involved in what can now be routinely achieved.
The most important aspect of Alexey's work is the interaction between mitoribosome and OXA1L and the associated flexibility. The fact that mitoribosome is flexible as such is not novel, but the particular flexibility associated with OXA1L interaction is. This is important for synthesis of membrane proteins, including respiratory chain proteins. Overall, this work significantly widens our understanding how mitoribosome functions. The work by Alexey Amunts lab resolves another mystery about basic biological processes necessary for creating life as we know it."
The sequencing of the human mitochondrial genome 40 years ago was a turning point in mitochondrial research, postulating a putative specialized mechanism for the synthesis of the mitochondrial transmembrane proteins. Indeed, the discovered gating mechanism of the human mitoribosome represents a unique occurrence. Therefore, the structural data offer a fundamental understanding into how bioenergetic proteins are synthesized in our body.
News Source: www.sciencedaily.com/
Article source: Yuzuru Itoh, Juni Andréll, Austin Choi, Uwe Richter, Priyanka Maiti, Robert B. Best, Antoni Barrientos, Brendan J. Battersby, Alexey Amunts. Mechanism of membrane-tethered mitochondrial protein synthesis. Science, 2021; 371 (6531): 846 DOI: 10.1126/science.abe0763
Cells burn more calories after just one bout of moderate aerobic exercise, OSU study finds
- Details
- Published on 03 May 2021

In a recent study testing the effects of exercise on overall metabolism, researchers at Oregon State University found that even a single session of moderate aerobic exercise makes a difference in the cells of otherwise sedentary people.
Mitochondria are the part of the cell responsible for the biological process of respiration, which turns fuels such as sugars and fats into energy, so the researchers focused only on mitochondria function.
"What we found is that, regardless of what fuel the mitochondria were using, there were mild increases in the ability to burn off the fuels," said Matt Robinson, lead author on the study and an assistant professor in the College of Public Health and Human Sciences.
OSU researchers recruited participants who do not follow a regular exercise routine and had them ride a stationary bike for an hour at a moderate intensity. They biopsied their muscles 15 minutes later to test how efficient the mitochondria were after the exercise was completed and compared those results with a resting day.
Post-exercise, study participants' mitochondria burned 12-13% more fat-based fuel and 14-17% more sugar-based fuel. While the effects were not drastic, they were consistent, Robinson said.
"It's pretty remarkable that even after just one hour of exercise, these people were able to burn off a little more fuel," he said.
Previous research in the field has long established that regular exercise creates lasting change in people's metabolism, making their bodies burn more energy even when they're not working out.
Prior studies have looked at highly trained or athletic people, but Robinson's team wanted to look specifically at singular bouts of exercise in people who were generally active and disease-free but who did not have structured exercise regimes. These people were on the lower end of fitness, which is associated with low mitochondrial abundance and energy production. Participants were monitored while working out at approximately 65% of their maximal effort, where they could keep up the cycling pace for an hour or more and still comfortably carry on a conversation.
Robinson said they're hoping these results help break down the mental barrier of people thinking they need to be elite athletes for exercise to make an impact on their health.
"From a big picture health perspective, it's very encouraging for people to realize that you can get health benefits from a single session of exercise," Robinson said. "We're trying to encourage people, 'You did one, why don't you try to do two? Let's do three.'
"We know that exercise is good for you, in general. But those benefits of that single bout of exercise seem to fade away after a day or two. You get the long-term benefits when you do that exercise again and again and you make it a regular habit."
In this study, Robinson's research team focused narrowly on mitochondria to find out how big a role mitochondria play in the overall function of muscle metabolism. Other studies are looking at changes in blood flow to the muscle and how the muscle metabolizes fats versus sugars.
From a disease perspective, Robinson said it's clear that obesity and diabetes involve impairments in metabolism. Physiologically, when the body undergoes exercise, sugars tend to be burned off first while fats are stored, but in cases of diabetes and obesity, there is some dysregulation in metabolism that causes the body to not be able to switch between the two types of fuel.
Exercise can help reset that system, he said.
"Since those get burned off in the mitochondria, our hope is that with exercise, we could increase the mitochondria and then improve how the body burns off fats and sugars," he said.
News source: www.sciencedaily.com
Article source: Sean A. Newsom, Harrison D. Stierwalt, Sarah E. Ehrlicher, Matthew M. Robinson. Substrate-specific Respiration of Isolated Skeletal Muscle Mitochondria after 1 h of Moderate Cycling in Sedentary Adults. Medicine & Science in Sports & Exercise, 2021; Publish Ahead of Print DOI: 10.1249/MSS.0000000000002615