Two Blood Markers of Schizophrenia Identified
- Details
- Published on 20 December 2021


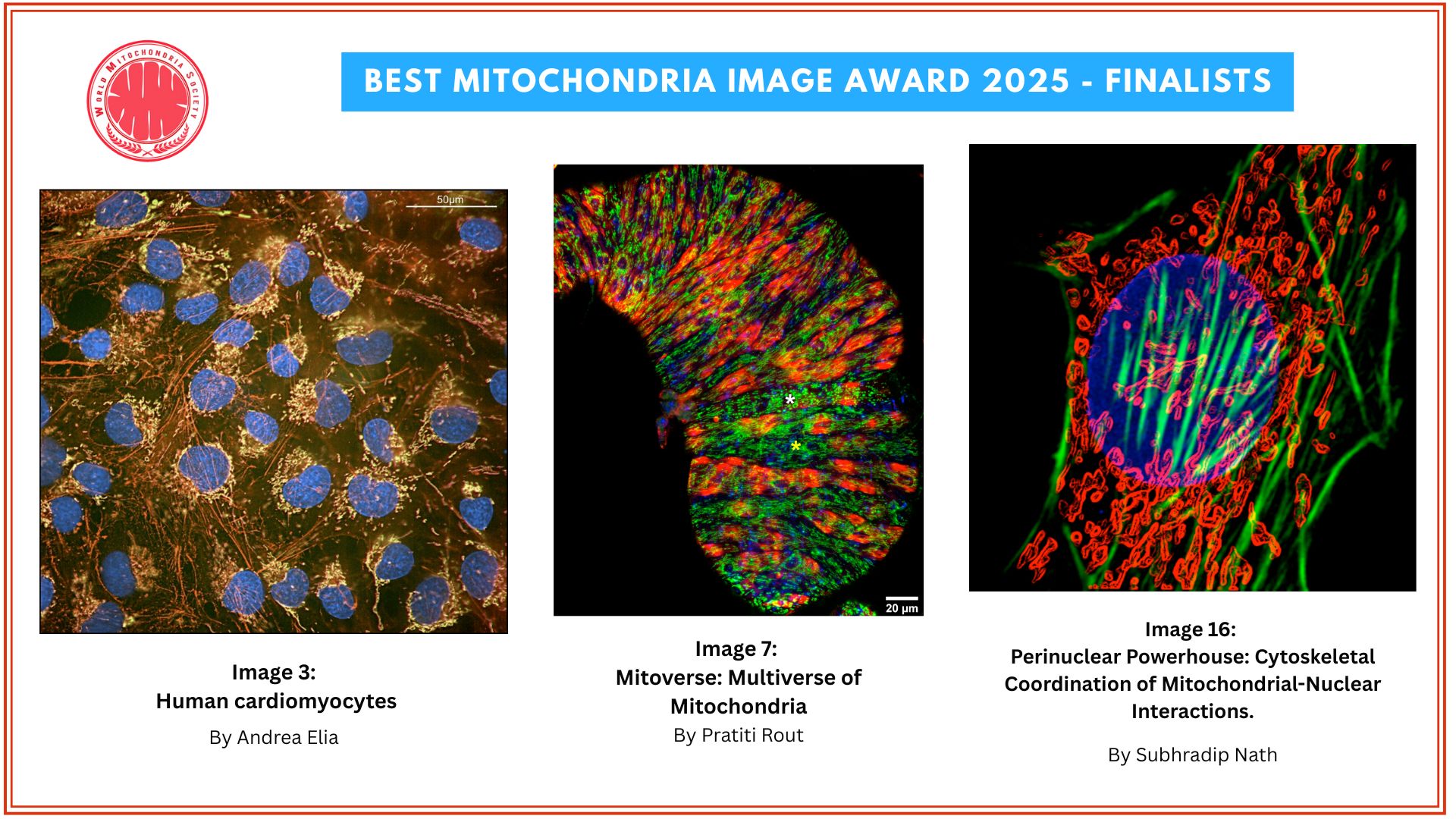
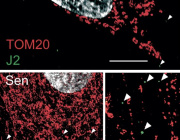
Mouse parvalbumin neurons. Credit: @UNIL/CHUV Inès Khadimallah
This phenomenal research by Synapsy brings us closer to diagnosing Schizophrenia by blood, and treating it.
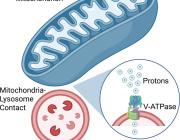
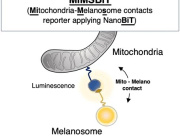
A research team from Synapsy has shown that the severity of the clinical symptoms of schizophrenia is strongly linked to blood biomarkers related to the deregulation of neuronal mitochondria.
A study conducted at the Centre for Psychiatric Neuroscience of the Lausanne University (UNIL) and the Lausanne University Hospital (CHUV), and supported by the National Centre of Competences in Research Synapsy (Synapsy), has shown, in an animal model, that the cellular mechanism for recycling mitochondria is deficient in parvalbumin neurons.
Mitochondria are generally able to eliminate their damaged parts by splitting, using a mechanism called mitophagy. This process involves a series of molecules whose production is controlled by miR137, a microRNA that plays a key role in their regulation. Inès Khadimallah, in collaboration with her colleagues, succeeded in demonstrating that the level of miR-137 was very high in the model, as was oxidative stress. In parallel, an element of cellular respiration expressed specifically by parvalbumin neurons, the COX6A2 molecule is decreased. "In other words, the mitochondria of parvalbumin neurons are dysfunctional following the increase in oxidative stress, and it can be shown through analyzing the levels of miR-137 and COX6A2 in the blood".
In their attempts to intervene directly on the free radicals produced by mitochondria, the neuroscientists showed, in the animal model, that the alterations of these two molecules, miR137 and COX6A2, can be completely corrected by an antioxidant compound that specifically targets mitochondria, called MitoQ.
Keep on track with us and stay updated with all the latest research!
A newly discovered anti-senescence function of Vitamin B2
- Details
- Published on 14 December 2021
A new study conducted by Kobe University will take your understanding of cell aging to a whole new level.
Japan is a super-aged society and as a result, research into aging is becoming increasingly important to resolve the accompanying medical and welfare issues, and to help people live healthily for longer.
Furthermore, it has been discovered that it is possible to prevent or ameliorate age-related disorders that occur more frequently as people get older, such as cancer, cardiovascular disease, Alzheimer’s and diabetes, by preventing the accumulation of senescent cells.
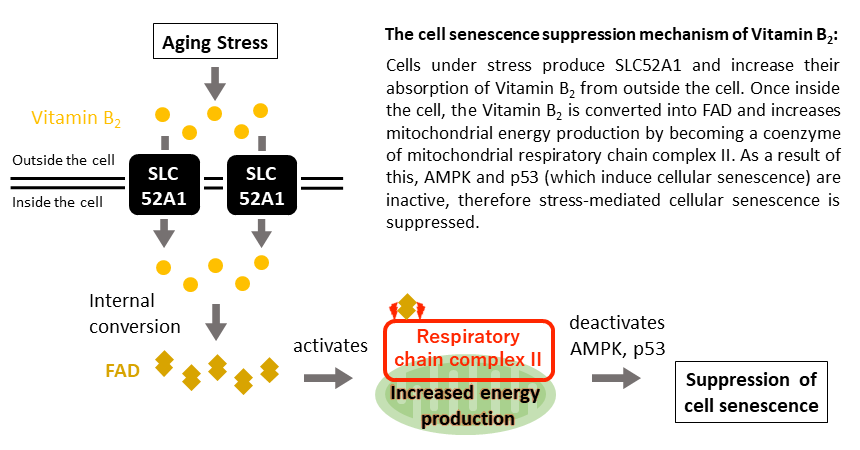
A group of Kobe University researchers have revealed that adding Vitamin B2 to cells that have been exposed to aging stress increases the mitochondria’s ability to produce energy and prevents cell aging.
The research team discovered a phenomenon whereby resistance to cellular senescence occurred as a result of increasing the amount of SLC52A1 produced. SLC52A1 is the protein responsible for transporting vitamin B2 into cells (vitamin B2 transporter*1).
Inside the cell, vitamin B2 is converted into a substance called Flavin Adenine Dinucleotide (FAD), a coenzyme*2 that promotes the chemical reactions necessary for biological activities such as energy production.
Stay tuned to keep updated about the indespensable role and potential of mitochondria.
Read more on this research here.
The mitochondrial protein Opa1 promotes adipocyte browning that is dependent on urea cycle metabolites
- Details
- Published on 09 December 2021
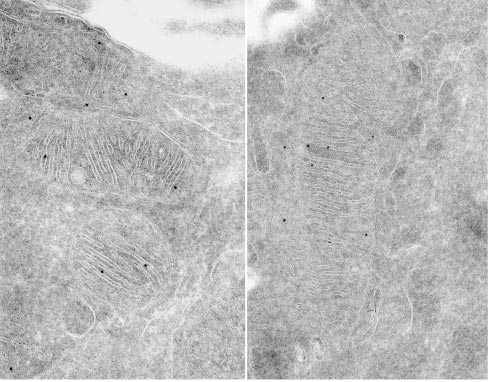
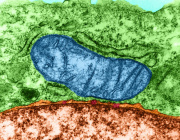
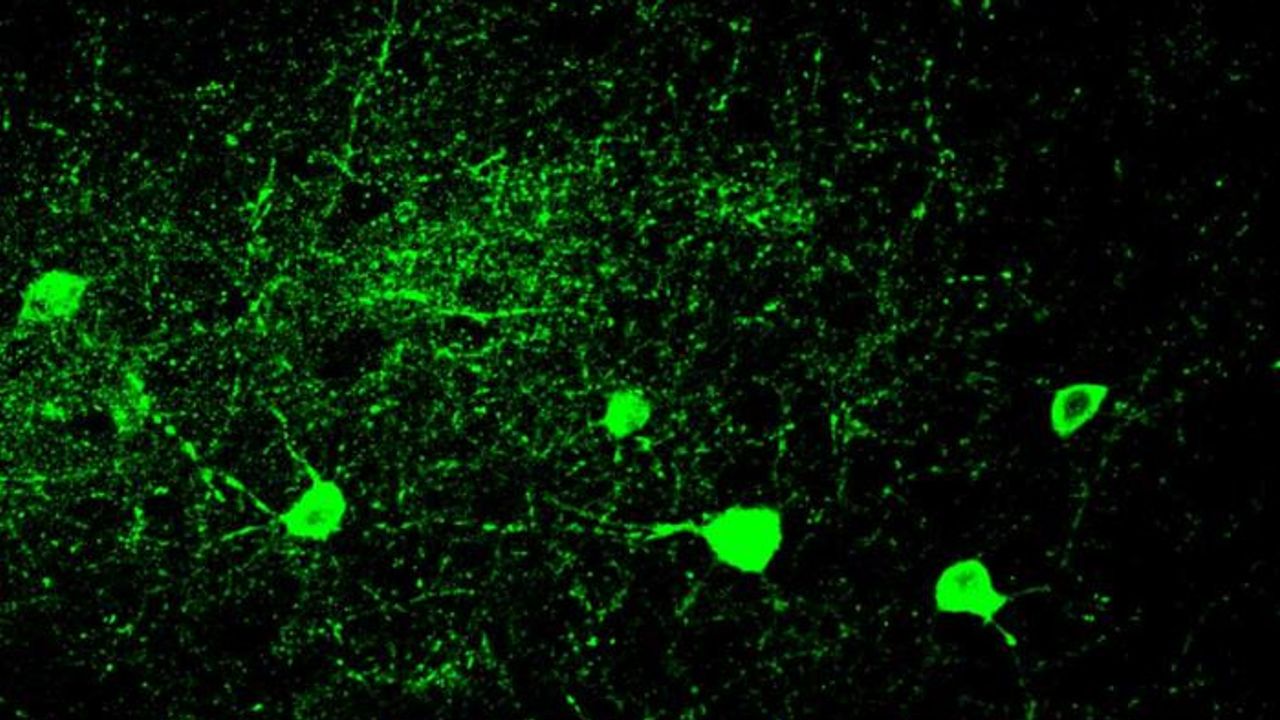
Submitochondrial localization of Mgm1/OPA1 protein as determined by immuno-electron microscopy. Credits: Loss of the Intermembrane Space Protein Mgm1/OPA1 Induces Swelling and Localized Constrictions along the Lengths of Mitochondria - Scientific Figure on ResearchGate.
The conversion of white to brown/beige adipocytes is a possible therapeutic strategy for tackling the current obesity epidemics. Mitochondria are known to be key for energy dissipation in brown fat, but it is unknown if they can drive adipocyte browning. In this study, they showed that the mitochondrial cristae biogenesis protein optic atrophy 1 (Opa1) facilitates cell-autonomous adipocyte browning.
They reported that, Adipose tissue OPA1 levels were reduced in two cohorts of patients with obesity. Also, the overexpression of Opa1 in mouse favored white adipose tissue expandability as well as browning, improving glucose tolerance and insulin sensitivity.
They identified, using transcriptomics and metabolomics analyses, Jumanji family chromatin remodeling protein Kdm3a and urea cycle metabolites, including fumarate, as effectors of Opa1-dependent browning.
They indicated, using flux analyses, that Opa1-dependent fumarate accumulation is dependent on the urea cycle.
In conclusion, they stated that the urea cycle links the mitochondrial dynamics protein Opa1 to white adipocyte browning.
Authors: Bean, C., Audano, M., Varanita, T. et al.
Defense or repair: How immune cells are controlled during wound healing
- Details
- Published on 09 December 2021

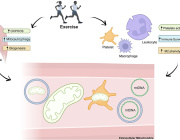
A Cologne-based research team has discovered that the metabolism of mitochondria, the energy suppliers of cells, in macrophages coordinate wound healing to a significant degree. Macrophages belong to the white blood cells and are also known as scavenger cells. Professor Dr. Sabine Eming and her collaborators and colleagues at the CECAD Cluster of Excellence for Aging Research at the University of Cologne showed that wound macrophages undergo different metabolic programs during tissue repair, which are required to support the successive phases for skin reconstruction after injury.
For more information press here.
Full article link: http://dx.doi.org/10.1016/j.cmet.2021.10.004
Image source:Hand photo created by shayne_ch13 - www.freepik.com
How some tissues can “breathe” without oxygen
- Details
- Published on 08 December 2021
Humans need oxygen molecules for a process called cellular respiration, which takes place in our cells’ mitochondria. Through a series of reactions called the electron transport chain, electrons are passed along in a sort of cellular relay race, allowing the cell to create ATP, the molecule that gives our cells energy to complete their vital functions.
In the past, however, scientists have noticed that cells are able to maintain some functions of the electron transport chain, even in the absence of oxygen. “This indicated that mitochondria could actually have partial function, even when oxygen is not the electron acceptor,” said Whitehead Institute postdoctoral researcher Jessica Spinelli. “We wanted to understand, how does this work? How are mitochondria capable of maintaining these electron inputs when oxygen is not the terminal electron acceptor?”
In a paper published December 2 in the journal Science, Whitehead Institute scientists and collaborators led by Spinelli have found the answer to these questions. Their research shows that when cells are deprived of oxygen, another molecule called fumarate can step in and serve as a terminal electron acceptor to enable mitochondrial function in this environment. The research, which was completed in the laboratory of former Whitehead Member David Sabatini, answers a long-standing mystery in the field of cellular metabolism, and could potentially inform research into diseases that cause low oxygen levels in tissues, including ischemia, diabetes and cancer.
The World Mitochondria Society will highlight this topic in the "13th Targeting Mitochondria Congress 2022".
View full article here.
More Articles...
- Cancer Cells Strengthen by Sucking Mitochondria out of Immune Cells Using ‘Tiny Tentacles’, Study Finds
- Scientists Explore the Creation of Artificial Organelles
- An Overactive Sweet Tooth May Spell Trouble for Our Cellular Powerplants
- Comprehensive Multi-Omics Analysis Reveals Mitochondrial Stress as a Central Biological Hub for Spaceflight Impact