Cancer Cells Strengthen by Sucking Mitochondria out of Immune Cells Using ‘Tiny Tentacles’, Study Finds
- Details
- Published on 22 November 2021


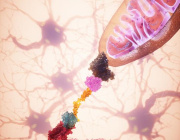
Cancer cells gain strength by forming “tiny tentacles” that suck the power out of immune cells, a recent study that could help develop new drug targets against the malignant disease has suggested.
Credits to National Cancer Institute
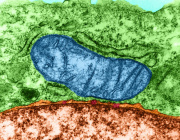
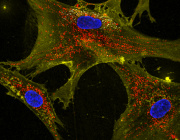
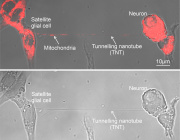
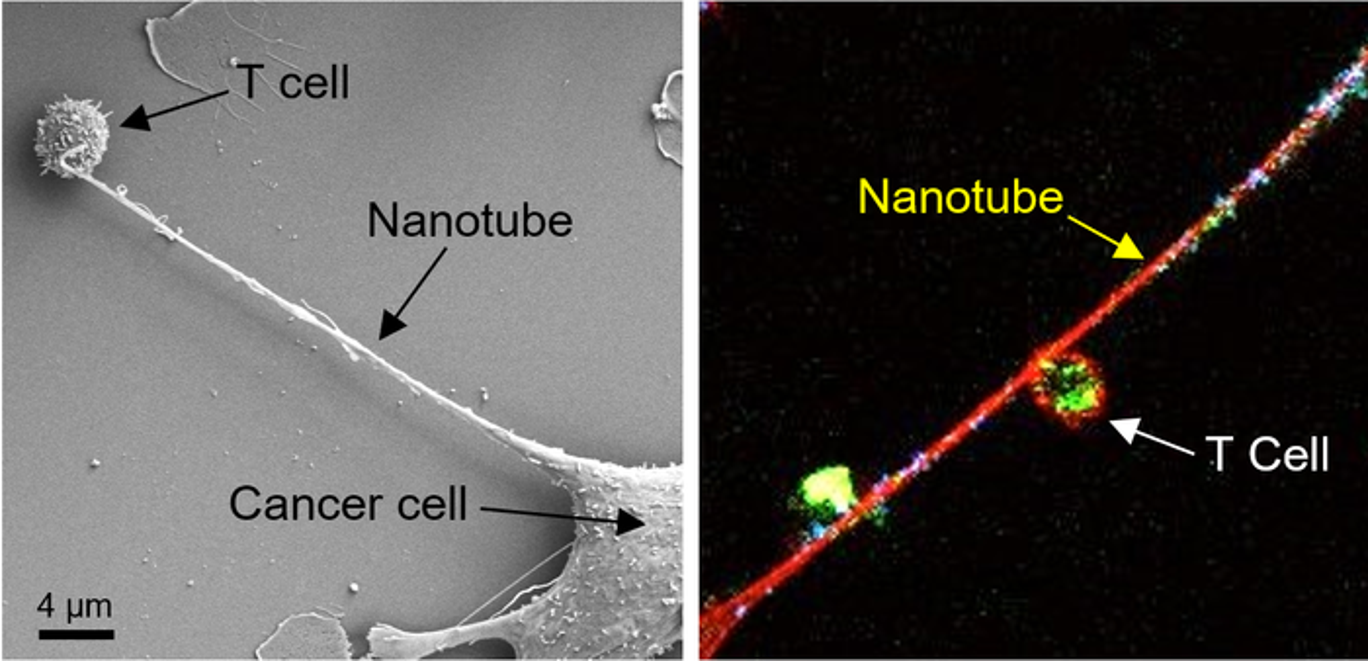
Left: Microscopic image shows formation of a nanotube between a breast cancer cell and an immune cell.
Right: Image shows mitochondria (labeled with green fluorescence dye) traveling from a T cell to a cancer cell through the intercellular nanotube.
Scientists from Brigham and Women's Hospital and MIT employed nanotechnology to identify a novel method that cancer may neutralize its would-be cellular adversaries. Immune cells are depleted, and cancer cells are boosted by slurping out the immune cell's mitochondria. Research published in Nature Nanotechnology suggests that the next generation of cancer immunotherapy may have a different target.
Shiladitya Sengupta, the corresponding author of the study and co-director of the Brigham's Center for Engineered Therapeutics, stated, "Cancer kills when the immune system is inhibited, and cancer cells are allowed to spread, and it looks that nanotube may help them accomplish both." When it comes to cancer cells evading the immune system, "this is an entirely new process, and it provides us with a new target to pursue. "Sengupta and colleagues co-cultured breast cancer cells with immune cells such as T cells to study the nanoscale interactions between cancer cells and immune cells. They noticed something strange when they used field-emission scanning electron microscopy: Immune cells and cancer cells seemed to be physically linked by thin tendrils, with diameters ranging from 100 to 1000 nanometers. Some of the nanotubes formed thicker tubes as they came together. As a result, they used a fluorescent dye to label mitochondria from T cells. They observed that the mitochondria were sucked out of the immune cells and delivered to cancer cells through the nanotube system.
According to co-corresponding author Hae Lin Jang, Ph.D., a principal investigator at the Center for Engineered Therapeutics, "by carefully preserving the cell culture condition and observing intracellular structures, we saw these delicate nanotubes, and they were stealing the energy source of the immune cells." In cancer cells, this type of activity has never been seen previously. When working with nanotubes, which are pretty delicate, we had to be extremely careful with the cells to avoid damaging them.The scientists next investigated what would happen if they blocked the cancer cells from gaining access to mitochondria. Using mice models of lung and breast cancer, scientists found that tumor development was significantly reduced when an inhibitor of nanotube production was administered.
"Finding combinations of medicines that potentially enhance outcomes is one of the aims of cancer immunotherapy," said lead author Tanmoy Saha, Ph.D., a postdoctoral researcher at the Center for Engineered Therapeutics. "Based on our findings, it seems that a nanotube formation inhibitor might be paired with cancer immunotherapies and examined to determine whether it improves patient outcomes."
Reference:
Saha, T., Dash, C., Jayabalan, R. et al. Intercellular nanotubes mediate mitochondrial trafficking between cancer and immune cells. Nat. Nanotechnol. (2021).
https://doi.org/10.1038/s41565-021-01000-4
Scientists Explore the Creation of Artificial Organelles
- Details
- Published on 23 September 2021
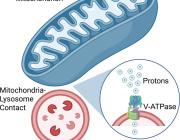
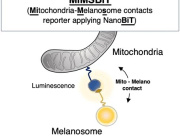
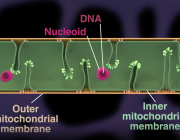
The human body is made of numerous different types of cells, which have small compartments known as organelles to perform complex biochemical reactions. These compartments have multiple enzymes that work together to execute important cellular functions. Researchers at the Center for Soft and Living Matter within the Institute for Basic Science (IBS, South Korea) have successfully mimicked these Nano spatial compartments to create ‘artificial mitochondria’ in the latest research published in Nature Catalysis as a cover article. They state the technology can be used to construct artificial organelles that can supply ATP or other useful molecules to cells in damaged or diseased tissues.
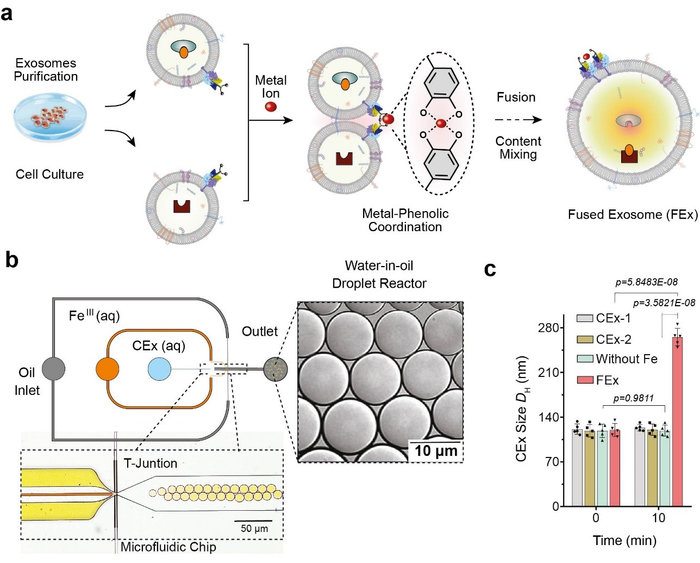 Credits to: Institute for Basic Science
Credits to: Institute for Basic Science
This was achieved through reprogramming of ‘exosomes’, which are small vesicles (diameter ~120 nm) that cells use for intercellular signaling. The researchers carried out the experiments using microfluidic droplet reactors, which generated small droplets that were of similar size as typical cells. (diameter ~10 μm) The researchers first aimed to facilitate controlled fusion of these exosomes within the droplets while preventing unwanted fusions.
They accomplished this by tailoring the exosome surfaces with molecules called catechol, which is a chelating agent that forms complexes with metal ions. This was in turn done by attaching the catechol onto antibodies that target specific cell markers, such as CD9. The complex-forming property of catechol allows them to drive fusions between exosomes when they are mixed with metal ions such as Fe3+. The membrane fusion occurs when the catechols on the surfaces bind to the iron and bring the vesicles to close proximity to one another.
News Source: https://scienmag.com/scientists-explore-the-creation-of-artificial-organelles/
DOI: 10.1038/s41929-021-00669-z
Targeting Mitochondria 2021 Congress
October 27-29, 2021
Interactive Online Congress
www.targeting-mitochondria.com
Comprehensive Multi-Omics Analysis Reveals Mitochondrial Stress as a Central Biological Hub for Spaceflight Impact
- Details
- Published on 04 August 2021
Spaceflight is known to impose changes on human physiology with unknown molecular etiologies.

To reveal these causes, the bioinformatician and principal investigator at KBR/NASA Ames Research Centera group, Dr. Afshin Beheshti, along with a group of scientists used a multi-omics, systems biology analytical approach using biomedical profiles from fifty-nine astronauts and data from NASA’s GeneLab derived from hundreds of samples flown in space to determine transcriptomic, proteomic, metabolomic, and epigenetic responses to spaceflight. According to the conducted research, overall pathway analyses on the multi-omics datasets showed significant enrichment for mitochondrial processes, as well as innate immunity, chronic inflammation, cell cycle, circadian rhythm, and olfactory functions.
However, NASA’s Twin Study provided a platform to confirm several of the researchers' principal findings.
In a nutshell, an intriguing finding happnened: they found evidence of altered mitochondrial function and DNA damage in the urine and blood metabolic data compiled from the astronaut cohort and NASA Twin Study data, indicating mitochondrial stress as a consistent phenotype of spaceflight.
DOI:https://doi.org/10.1016/j.cell.2020.11.002
Click here to view the full article.
An Overactive Sweet Tooth May Spell Trouble for Our Cellular Powerplants
- Details
- Published on 06 August 2021
Recent researches demonstrated by Dr. Ning Wu, an assistant professor at Van Andel Institute, Grand Rapids, shows that the average American eats roughly 22 teaspoons of added sugar a day: more than three times the recommended amount for women and more than double the recommended amount for men.
Although this overconsumption is known to contribute to Type 2 diabetes and other disorders, the exact ways in which eating too much sugar sets the stage for metabolic diseases on a cellular level has been less clear.
A recent study led by Van Andel Institute scientists has found that surplus sugar may cause our cellular powerplants — called mitochondria— to become less efficient, reducing their energy outputs.
The findings, published today in Cell Reports, highlight the cellular implications of excessive sugar consumption and provide an important new model to study the initial metabolic events that may contribute to diabetes development.
“The body needs sugar, or glucose, to survive, but, as the saying goes: ‘All good things in moderation,’” said Dr. Ning Wu. “We found that too much glucose in cells, which is directly linked to the amount of sugar consumed in one’s diet, affects lipid composition throughout the body, which in turn affects the integrity of mitochondria. The overall effect is a loss of optimal function.”
Using their new model, Wu and her colleagues demonstrated that excess glucose reduces the concentration of polyunsaturated fatty acids (PUFAs) in the mitochondrial membrane and makes mitochondria less efficient. PUFAs are vital players in supporting mitochondrial function and mediating a host of other biological processes such as inflammation, blood pressure and cellular communication.
Instead, excess glucose is synthesized into a different form of fatty acid that isn’t as efficient or as flexible as PUFAs. This upends the lipid composition of the membrane and puts stress on the mitochondria, damaging them and impacting their performance.

Picture representing the study on mouses.
Wu and her colleagues were able to reverse this detrimental effect by feeding their mouse models a low-sugar ketogenic diet, which suggests that reducing glucose and restoring normal membrane lipid composition supports healthy mitochondrial integrity and function. They also found that consuming excess carbohydrates reduces the beneficial effect of PUFA supplements.
To conclude, Dr. Wu says: “Although we may not always notice the difference in mitochondrial performance right away, our bodies do,” Wu explained. “If the lipid balance is thrown off for long enough, we may begin to feel subtle changes, such as tiring more quickly. While our study does not offer medical recommendations, it does illuminate the early stages of metabolic disease and provides insights that may shape future prevention and therapeutic efforts.”
News Source: https://www.vai.org/ning-wu-sugar-mitochondria/
View full article here.
UCPH Researchers Prove Powerhouse Malfunction as the Major Cause of Parkinson’s Disease
- Details
- Published on 12 July 2021
UCPH Researchers Prove Powerhouse Malfunction as the Major Cause of Parkinson’s Disease
RESEARCH: The major cause of Parkinson’s Disease is a dysregulation of immune genes central for fighting against viruses, a new study reveals. Researchers from the University of Copenhagen show that this dysregulation leads to a malfunction in the cell’s powerhouse, which cannot produce sufficient energy for neurons to stay alive, causing them to gradually die.

Photo: Colourbox.
12,000 people in Denmark and 7 to 10 million people worldwide suffer from Parkinson’s Disease (PD). It is the second most common neurogenerative disorder of aging and the most common movement disorder, but the cause of the disease is largely unknown.
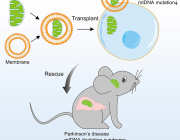
In a new study, researchers from the University of Copenhagen show that the most common form of the disease, encompassing 90 to 95 percent of all Parkinson’s Disease cases known as sporadic PD, is caused by a blockage of a pathway that regulates the nerve cell’s powerhouse, the mitochondria.
‘Just like when people eat, cells take what they need and get rid of the rest waste products. But if our brain cells have this specific kind of signaling blockage, it means that the powerhouse of the cell – mitochondria – cannot get cleaned up after being damaged’, explains corresponding author and group leader Professor Shohreh Issazadeh-Navikas at the Biotech Research & Innovation Centre.
The blockage leads to an accumulation of high amounts of damaged mitochondria, while not being able to produce enough energy for the cells. It causes neurons to gradually die, which is the reason for the development of Parkinson’s Disease symptoms, and why it leads to dementia.
The blockage is caused by a dysregulation of the immune genes, more specifically a pathway called type 1 interferon, which is normally important for fight against viruses, but now we show that it is also responsible for regulating the energy supply of the nerve cells.
‘Every part of our body needs to be regulated. We get a signal to stop eating, when we are full, and the same thing happens everywhere else in our body. If we get an infection, parts of our body need to fight it and stop it from replicating. But when the infection is cleaned up, the signal should subside. This is the job of a protein called PIAS2. That causes the blockage of the type 1 interferon-pathway, and when the infection is over, the blockage should stop and go back to normal. But that does not seem to be the case in patients with Parkinson’s Disease. We further demonstrate that this dysregulation leads to a defect in the mitochondrial energy supply, as mentioned before’, says Shohreh Issazadeh-Navikas.
These pathways are very important for brain functions, but they are also associated with microbial and virus recognition. For example, they are very important for fighting COVID-19, and a mutation in the related gene has been shown to be linked to a deadly outcome after contracting COVID-19.
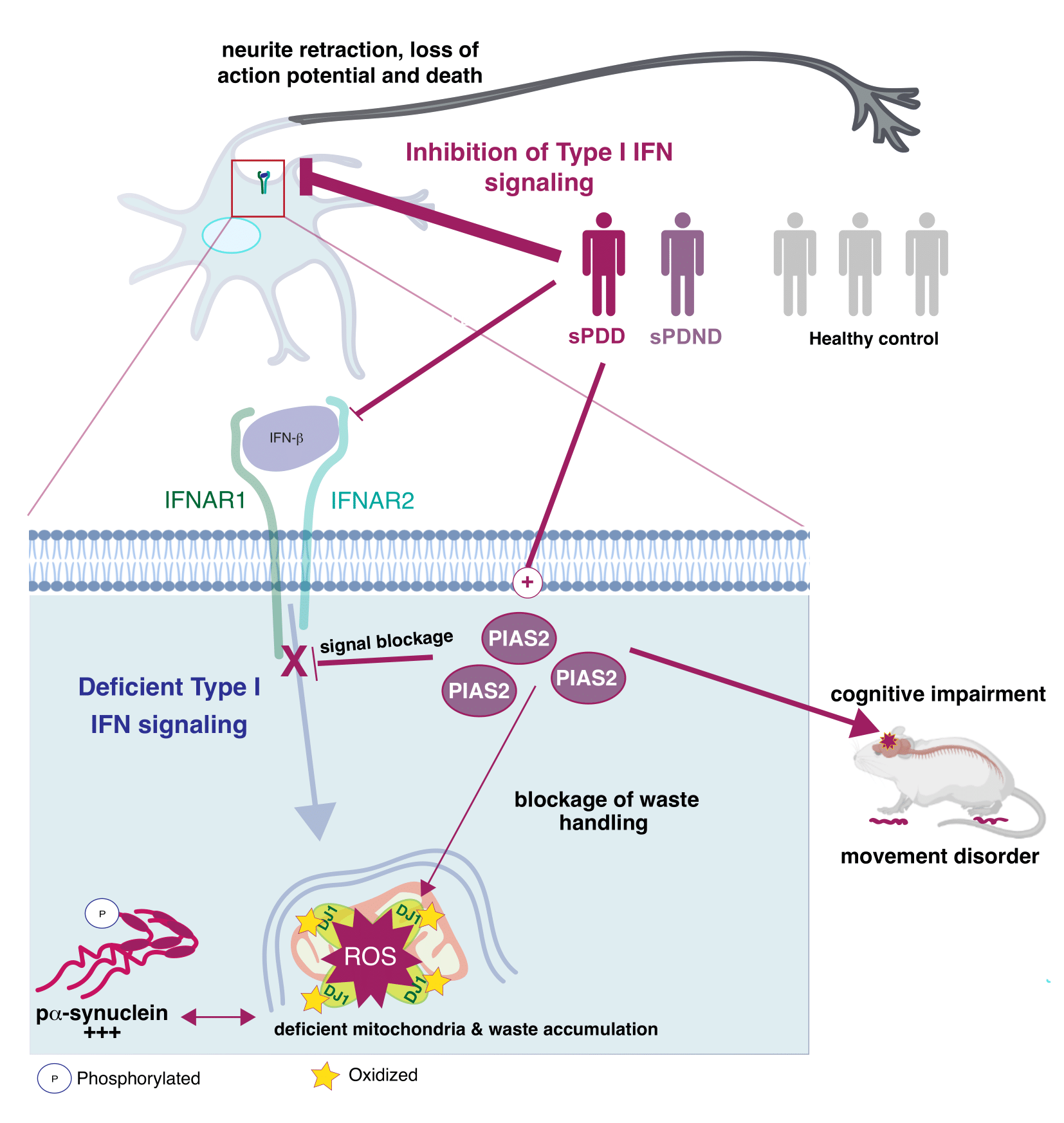
A detail look into what happens in the neurons. (See Illustration Above)
Combining Datasets for a Bigger Picture
The researchers combined and analyzed four data sets, which studied neurons from brains with Parkinson’s Disease and looked at what type of genes they express.
They then looked at which gene patterns were disturbed in patients with Parkinson’s Disease and especially those who had also developed PD with dementia.
In order to test the results, the major findings of the combined data was tried in three different mouse models using a negative regulator of the type I interferon pathway, PIAS2, which had been identified from the patients study as one of the key proteins linked to the progression of Parkinson’s Disease and dementia.
‘We show that a high accumulation of the PIAS2-protein is what is causing the blockage in the pathway, which should have activated the processes responsible for removing damaged protein and mitochondrial garbage’, says Shohreh Issazadeh-Navikas.
‘The accumulation of damaged mitochondrial mass further leads to increase of other toxic proteins. So when we compare patients to same-aged healthy patients without Parkinson’s Disease, we see that this PIAS2-protein is highly expressed in the neurons, which is why this pathway should be evaluated for potential roles in the other forms of familial Parkinson’s Disease that we have not studied here.’
The researchers hope the study will encourage research to counteract the pathway blockage, which could have a beneficial impact on the disease and towards preventing dementia.
In the next stages, the Shohreh Group will study how the pathway contributes to neuronal homeostasis and survival, as well as how its dysregulation causes neuronal cell death.
News Source: University of Copenhagen
More Articles...
- It’s True: Stress Does Turn Hair Gray (And It’s Reversible)
- Age-Related Mitochondrial Dysfunction as a Key Factor in COVID-19 Disease
- New warning system discovered in the immune defence
- Mitochondrial Functioning and the Relations among Health, Cognition, and Aging: Where Cell Biology Meets Cognitive Science