It’s True: Stress Does Turn Hair Gray (And It’s Reversible)
- Details
- Published on 02 July 2021



Legend has it that Marie Antoinette’s hair turned gray overnight just before her beheading in 1791.
Though the legend is inaccurate—hair that has already grown out of the follicle does not change color—a new study(link is external and opens in a new window) from researchers at Columbia University Vagelos College of Physicians and Surgeons is the first to offer quantitative evidence linking psychological stress to graying hair in people.
And while it may seem intuitive that stress can accelerate graying, the researchers were surprised to discover that hair color can be restored when stress is eliminated, a finding that contrasts with a recent study in mice that suggested that stressed-induced gray hairs are permanent.
“Understanding the mechanisms that allow ‘old’ gray hairs to return to their ‘young’ pigmented states could yield new clues about the malleability of human aging in general and how it is influenced by stress,” Picard says.
“Our data add to a growing body of evidence demonstrating that human aging is not a linear, fixed biological process but may, at least in part, be halted or even temporarily reversed.”
Studying hair as an avenue to investigate aging
“Just as the rings in a tree trunk hold information about past decades in the life of a tree, our hair contains information about our biological history,” Picard says. “When hairs are still under the skin as follicles, they are subject to the influence of stress hormones and other things happening in our mind and body. Once hairs grow out of the scalp, they harden and permanently crystallize these exposures into a stable form.”
Though people have long believed that psychological stress can accelerate gray hair, scientists have debated the connection due to the lack of sensitive methods that can precisely correlate times of stress with hair pigmentation at a single-follicle level.
Splitting hairs to document hair pigmentation
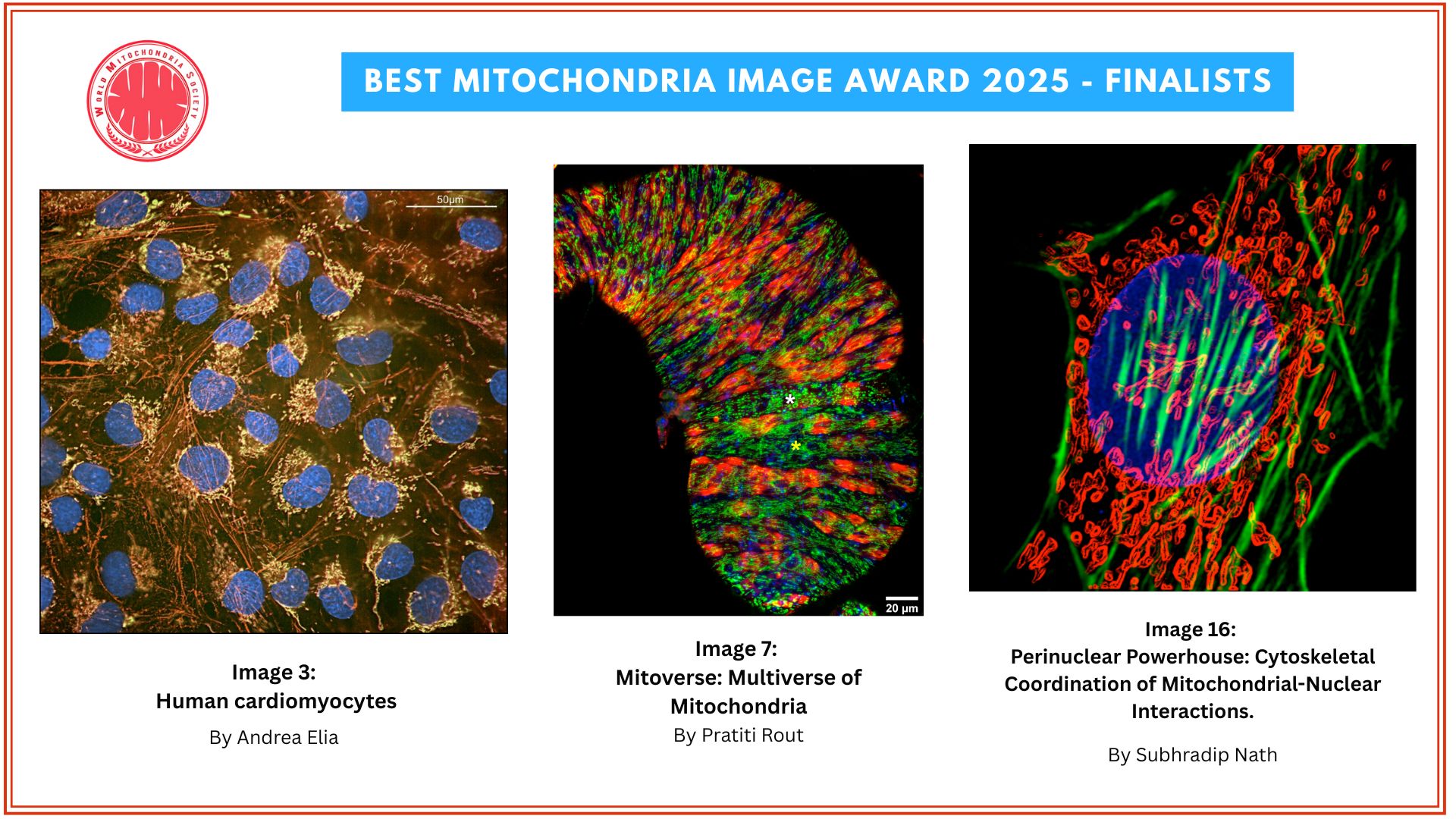
Ayelet Rosenberg, first author on the study and a student in Picard’s laboratory, developed a new method for capturing highly detailed images of tiny slices of human hairs to quantify the extent of pigment loss (graying) in each of those slices. Each slice, about 1/20th of a millimeter wide, represents about an hour of hair growth.
“If you use your eyes to look at a hair, it will seem like it’s the same color throughout unless there is a major transition,” Picard says. “Under a high-resolution scanner, you see small, subtle variations in color, and that’s what we’re measuring.”
 Hair pigmentation patterns of 100 hairs from a male and female study participant. Darker hair colors represented in red; lighter in blue. Image from Rosenberg et al. (2021).
Hair pigmentation patterns of 100 hairs from a male and female study participant. Darker hair colors represented in red; lighter in blue. Image from Rosenberg et al. (2021).
The researchers analyzed individual hairs from 14 volunteers. The results were compared with each volunteer’s stress diary, in which individuals were asked to review their calendars and rate each week’s level of stress.
The investigators immediately noticed that some gray hairs naturally regain their original color, which had never been quantitatively documented, Picard says.
When hairs were aligned with stress diaries by Shannon Rausser, second author on the paper and a student in Picard’s laboratory, striking associations between stress and hair graying were revealed and, in some cases, a reversal of graying with the lifting of stress.
“There was one individual who went on vacation, and five hairs on that person’s head reverted back to dark during the vacation, synchronized in time,” Picard says.
Blame the mind-mitochondria connection
To better understand how stress causes gray hair, the researchers also measured levels of thousands of proteins in the hairs and how protein levels changed over the length of each hair.
Changes in 300 proteins occurred when hair color changed, and the researchers developed a mathematical model that suggests stress-induced changes in mitochondria may explain how stress turns hair gray.
“We often hear that the mitochondria are the powerhouses of the cell, but that’s not the only role they play,” Picard says. “Mitochondria are actually like little antennas inside the cell that respond to a number of different signals, including psychological stress.”
The mitochondria connection between stress and hair color differs from that discovered in a recent study of mice, which found that stress-induced graying was caused by an irreversible loss of stem cells in the hair follicle.
“Our data show that graying is reversible in people, which implicates a different mechanism,” says co-author Ralf Paus, PhD, professor of dermatology at the University of Miami Miller School of Medicine. “Mice have very different hair follicle biology, and this may be an instance where findings in mice don’t translate well to people.”
Hair re-pigmentation only possible for some
Reducing stress in your life is a good goal, but it won’t necessarily turn your hair to a normal color.
“Based on our mathematical modeling, we think hair needs to reach a threshold before it turns gray,” Picard says. “In middle age, when the hair is near that threshold because of biological age and other factors, stress will push it over the threshold and it transitions to gray.
“But we don’t think that reducing stress in a 70-year-old who’s been gray for years will darken their hair or increasing stress in a 10-year-old will be enough to tip their hair over the gray threshold.”
News source: www.cuimc.columbia.edu/
Age-Related Mitochondrial Dysfunction as a Key Factor in COVID-19 Disease
- Details
- Published on 08 June 2021
The Scientific Committee would like to share this excellent article written by Guillermo López-Lluch and al. on Age-Related Mitochondrial Dysfunction as a Key Factor in COVID-19 Disease. This article will be presented in Targeting Mitochondria Congress 2021.

The Scope is that Coronavirus Disease 2019 (COVID-19) is a complex respiratory and thrombogenic disease caused by a new strain of coronavirus known as Severe Acute Respiratory Syndrome-Coronavirus 2 (SARS-CoV-2). Many of the patients infected with SARS-CoV-2 are asymptomatic or show low intensity symptoms. However, around 20% of the patients, mainly elderly people, manifest severe symptoms and high mortality ratio. COVID-19 predominant symptoms are high fever, intense cough, pneumonia and myalgia, mainly leading to acute respiratory distress syndrome (ARDS). Diarrhea and nausea can precede fever or respiratory symptoms. These symptoms can be aggravated in senior patients aged more than 70 years.
To conclude, we postulate here that the maintenance of mitochondrial health through healthy life habits, pharmacological compounds o dietary supplements able to improve mitochondrial activity, dynamics and turnover would reduce the levels of inflammatory cytokines and the severity of COVID-19 and other respiratory diseases such as seasonal flu. These and other “geroprotective” treatments must be used to treat or prevent COVID-19. Interventions and compounds able to improve mitochondrial health in the elderly would reduce or even eliminate the cytokine storm induced by SARS-CoV-2 infection and, therefore, reduce the grade of ARDS associated with COVID-19.
Article link: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7648491/
Targeting Mitochondria 2021 Congress
October 27-29, 2021
Berlin, Germany & Virtual Congress
www.targeting-mitochondria.com
Mitochondrial Functioning and the Relations among Health, Cognition, and Aging: Where Cell Biology Meets Cognitive Science
- Details
- Published on 26 May 2021
The Scientific Committee of the World Mitochondria Society would like to share this article by David C. Geary, Department of Psychological Sciences, University of Missouri on Mitochondria, general intelligence, health and aging.

Credit: https://www.mydocuc.com/health-aging/
The Scope is: Performance in one cognitive domain, such as attentional control, is positively correlated with performance in all other cognitive domains, such as reading comprehension, and performance in all of these domains is correlated with current and predictive of later health outcomes. These relations suggest a common biological mechanism that contributes to cognition and health; moreover, this mechanism has been linked to systematic and parallel declines in cognition and health with normal aging. Mitochondrial functioning, including contributions to cellular energy production, control of oxidative stress, immunity, and intracellular signaling (among others), is well situated to explain at least some of these links.
The Conclusion is: There is now consistent evidence that various mitochondrial functions, including energy production, control of oxidative stress, and intracellular signalling, among others, contribute to the well-documented relations between cognition, health, and aging. These relations provide a natural link between research in cell biology and cognitive science. Advances in the latter are potentially useful for the development of measures that will be the most sensitive to age- or disease-related disruptions of mitochondrial functions or therapeutic enhancement of these functions and their potential influence on cognition.
DOI: https://doi.org/10.3390/ijms22073562
Targeting Mitochondria 2021 Congress
October 27-29, 2021
Berlin, Germany & Virtual Congress
www.targeting-mitochondria.com
New warning system discovered in the immune defence
- Details
- Published on 26 May 2021
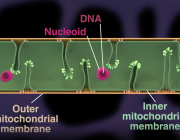
Researchers at Linköping University in Sweden have discovered a previously unknown warning system that contributes to the body's immune system. Mitochondria in the white blood cells secrete a web of DNA fibres that raises the alarm.
Credit: Reproduced with permission from Proceedings of the National Academy of Sciences USA
Introduction is: White blood cells are major components of the body’s immune defence, and the research group has shown that several types of these cells react against small DNA fragments that are similar to the DNA from bacteria and viruses. The white blood cells spray out a web consisting of mitochondrial DNA (mtDNA) strands. Mitochondria are present in all cells and normally produce the energy needed by the cell, by burning sugar and fat to form water and carbon dioxide.
The web that the mitochondria release sends signals to the surrounding cells that the body is under attack, and cause other white blood cells to release a signal substance known as “interferon type 1”. This substance helps the immune system to combat the infection.
Conclusion is: High levels of interferon type 1, the signal substance activated by the mtDNA webs, occur in several autoimmune diseases and several types of cancer. The researchers believe that it may be possible to quantify the secreted mtDNA molecules and interpret the warning signals, and in this way understand these diseases better.
Full Article: https://www.eurekalert.org/pub_releases/2018-01/lu-nws011218.php
Targeting Mitochondria 2021 Congress
October 27-29, 2021
Berlin, Germany & Virtual Congress
www.targeting-mitochondria.com
Mitochondria, general intelligence, health and aging
- Details
- Published on 26 May 2021
The Scientific committee would like to share this excellent article written by Eric Stann on Intelligence can link to health and aging.
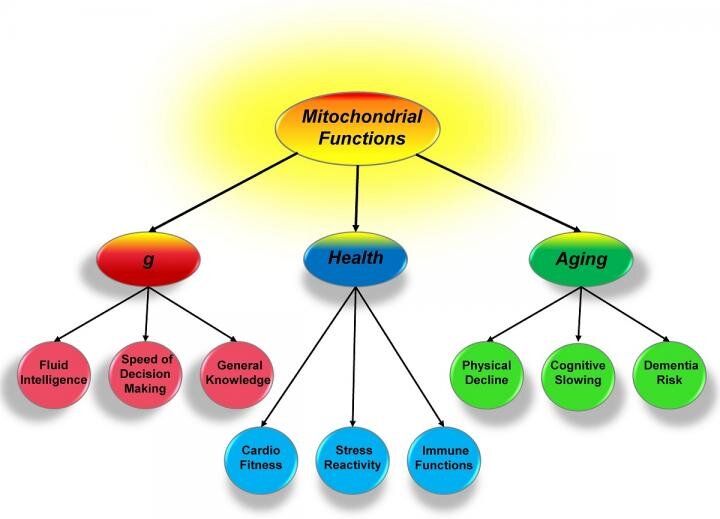
Credit: University of Missouri
As Introduction: For over 100 years, scientists have sought to understand what links a person's general intelligence, health and aging. In a new study, a University of Missouri scientist suggests a model where mitochondria, or small energy producing parts of cells, could form the basis of this link. This insight could provide valuable information to researchers studying various genetic and environmental influences and alternative therapies for age-related diseases, such as Alzheimer's disease.
To Conclude: These systems are being used over and over again, and eventually their heavy use results in gradual decline. Knowing this, we can help explain the parallel changes in cognition and health associated with aging. Also with good mitochondrial function, the aging processes will occur much more slowly. Mitochondria have been relatively overlooked in the past, but are now considered to relate to psychiatric health and neurological diseases.
Article link: https://www.eurekalert.org/pub_releases/2019-05/uom-icl050819.php
Targeting Mitochondria 2021 Congress
October 27-29, 2021
Berlin, Germany & Virtual Congress
www.targeting-mitochondria.com
More Articles...
- Big change from small player -- Mitochondria alter body metabolism and gene expression
- Parkinson, cancer, type 2 diabetes share a key element that drives disease
- Interaction of mitochondria and lysosomes key in Parkinson's disease
- Mitochondrial enzyme found to block cell death pathway points to new cancer treatment strategy