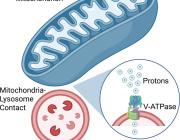
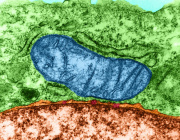
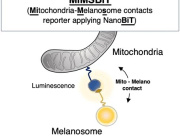
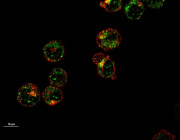
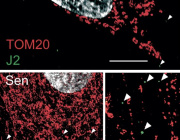
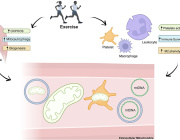
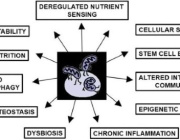
Pleiotropic Effects of Mitochondria in Aging
- Details
- Published on 22 March 2022


News Release, World Mitochondria Society, Berlin - Germany – March 22, 2022
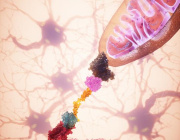
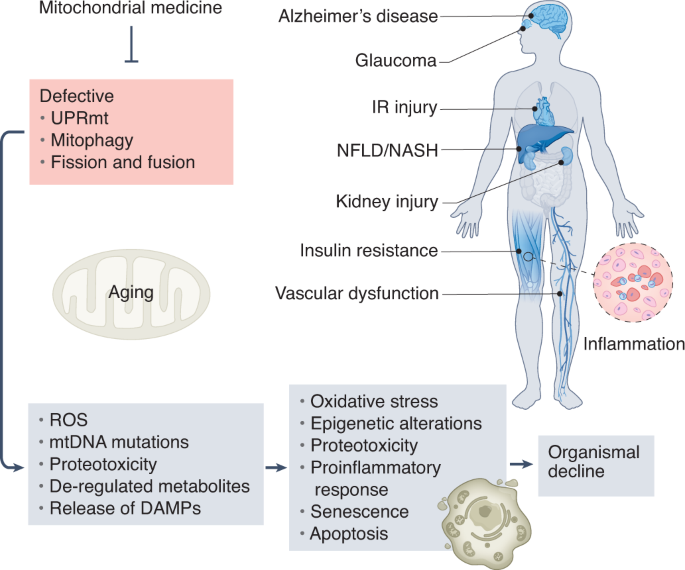
The mitochondrial stress-response (MSR) network contributes to the reconstitution of cellular homeostasis by preventing mitochondrial proteotoxicity and by redistributing and removing irreversibly damaged elements of the mitochondria. Recently, scientists have gained considerable insights into why a decline in the robustness of these MSR pathways contributes to cellular damage and organismal deterioration.
This study by Lima et al., published in nature aging, described the pleiotropic effects of mitochondrial dysfunction in aging.
Pleiotropic effects of mitochondria in aging
They outlined the major mitochondrial stress pathways, how their failure is interconnected with the expansion of mitochondrial DNA mutations and deregulated metabolism, and how this affects cellular and organismal homeostasis.
They furthermore provided an integrated map of how combined mitochondrial defects impact several features of aging, suggesting conserved links that could potentially be harnessed to slow the aging process. They described recent evidence arguing that defects in these conserved adaptive pathways contribute to aging and age-related diseases. Signaling pathways regulating the mitochondrial unfolded protein response, mitochondrial membrane dynamics, and mitophagy are discussed, emphasizing how their failure contributes to heteroplasmy and de-regulation of key metabolites.
The current understanding of how these processes are controlled and interconnected explains how mitochondria can widely impact fundamental aspects of aging.
Targeting Mitochondria 2022 will dedicate w whole session to Nuclear-Mitochondrial Interactions and their Effect on Longevity and Health. Professional speakers like Dr. Raghavan Pillai Raju will discuss the role of mitochondria in aging.
Don't miss out and register now.
Media contact:
World Mitochondria Society
This email address is being protected from spambots. You need JavaScript enabled to view it.
+33-1-5504-7755
Targeting Mitochondria 2022 Congress
October 26-28, 2022 - Berlin, Germany
wms-site.com
Exercise in Post-acute COVID-19 Syndrome Patients: Fatty Acid Oxidation & Lactate Production
- Details
- Published on 21 March 2022
News Release, World Mitochondria Society, Berlin - Germany – March 21, 2022
After acute infection with the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), many individuals experience a range of symptoms including dyspnea, exercise intolerance, and chest pain commonly referred to as “post–COVID-19 syndrome” or as post-acute sequelae of SARS-CoV-2 infection (PASC). Exertional dyspnea and physical activity intolerance in PASC can be debilitating despite mild acute coronavirus disease (COVID-19) and normal resting pulmonary physiology and cardiac function.
There is an urgent need to understand the pathogenesis of PASC and find effective treatments. The cardiopulmonary exercise test (CPET) is commonly used to investigate unexplained exertional dyspnea; as such, it could provide insight into mechanisms of PASC. CPET data can be used to calculate rates of β-oxidation of fatty acids (FATox) and of lactate clearance, providing insight into mitochondrial function. Fit individuals have better mitochondrial function and a higher rate of FATox during exercise than less fit individuals. In this study, they investigated whether patients with PASC had compromised mitochondrial function during graded exercise.
Data were obtained via retrospective review of the electronic medical record from a cohort of 50 subjects with PASC that were consecutively referred to and willing to complete CPET in the Pulmonary Physiology Laboratory at National Jewish Health between June 2020 and April 2021. Patients were assessed on a cycle ergometer using a continuous ramp protocol to exhaustion. Cardiovascular, ventilatory, metabolic, and gas exchange data were collected using a metabolic cart (Ultima Cardio2 System; Med graphics) per standard protocol. As a final step, patient data were compared with results from two published cohorts that included subjects tested with CPET in Denver, Colorado.
- Pulmonary function testing (PFT) showed mostly normal resting airflow and gas transfer capacity. All six patients (12%) with PFT abnormalities had preexisting illnesses, including asthma (n = 3) or interstitial lung disease. Resting transthoracic echocardiogram was obtained in 39 patients (78%) within 2 ± 3 months of CPET.
- Left ventricular systolic function was normal in all patients. Among the 50 patients who underwent CPET, the mean time from COVID-19 diagnosis to the CPET was 6 ± 4 months.
- Regardless of the presence of comorbidities, among the 39 patients with PASC who had arterial catheters in place, mean lactate was significantly higher; and in all 50 patients with PASC, calculated levels of FATox were significantly lower during exercise when compared with historical cohorts of subjects who are moderately active or with metabolic syndrome.
The data suggest that abnormally low FATox and altered lactate production by skeletal muscle as a putative cause of / contributor to the functional limitation of patients with PASC.
Normally, as glycolysis increases with exercise intensity, lactate is oxidized for fuel in mitochondria, mainly in adjacent slow-twitch muscle fibers. Like FATox, lactate clearance capacity is a useful surrogate for mitochondrial function. In patients with PASC, even in those with normal pre–COVID-19 physical fitness and free of comorbidities, the metabolic disturbances of the skeletal muscle during exercise may be worse than those reported in moderately active individuals or in individuals with metabolic syndrome. Whereas rising blood lactate levels are expected during high exercise intensity (as glycolytic flux exceeds the rate of mitochondrial pyruvate oxidation), a high blood lactate at lower exercise levels indicates mitochondrial dysfunction. The inappropriately high arterial lactate levels at relatively low exercise intensity in patients with PASC indicate that the transition from FATox to CHOox occurs prematurely, suggesting metabolic reprogramming and dysfunctional mitochondria.
This study provides the first evidence of mitochondrial dysfunction that advances our understanding of the pathogenesis of PACS in patients with preserved pulmonary and cardiac function. Future studies into the mechanisms of mitochondrial dysfunction in individuals with PACS will help accelerate the development of therapies to improve their functional status.
The implication of mitochondria in COVID-19 pathogenesis will be highlighted in Targeting Mitochondria 2022. If you have any studies that fit this title, you can submit your abstract now and share your work with the world mitochondria society.
© Image - kjpargeter, freepik
Media contact:
World Mitochondria Society
This email address is being protected from spambots. You need JavaScript enabled to view it.
+33-1-5504-7755
Targeting Mitochondria 2022 Congress
October 26-28, 2022 - Berlin, Germany
wms-site.com
The Alternate Version of Kreb's Cycle Underlying Cellular Identity
- Details
- Published on 16 March 2022
News Release, World Mitochondria Society, Berlin - Germany – March 16, 2022
Mitochondria and the Krebs cycle were discovered in the race to find causes and treatments for cancer and those pathways are learned by every student.
A new research, published in nature, that turned down the Krebs breakthrough that won him a Nobel Prize, says there is a complete alternative to this canonical cycle, also called the citric acid and tricarboxylic acid (TCA) cycle, with the downside the work is only in cells, not animals, so it remains exploratory.
Through several converging lines of evidence, the paper posits that an alternate version of the Krebs cycle takes place partly in the mitochondria and partly in the cytosol. Rather than burning sugar for energy, this alternate version allows cells to use the carbons in sugar to build important molecules such as lipids for cell membranes.
Not only that, but a cell’s use of one or the other version of the TCA cycle is associated with changes in its identity, the team found. The new results came out of a collaboration in the Finley lab between Gerstner Sloan Kettering graduate student Paige Arnold and Tri-Institutional MD-PhD student Benjamin Jackson. Arnold had been using carbon-tracing techniques to study the flow of carbons through the TCA cycle in different cell types. She had noticed, for example, that there seemed to be variation in the extent to which cells put their carbons into the TCA cycle versus skipping one part of it.
Around the same time, Jackson was using computational methods to analyze publicly available data from experiments in which the genome-editing tool CRISPR had been used to systematically knock out genes for various enzymes, one at a time, to see what effect this had on cells.
“You would hypothesize that if the TCA cycle were one functional module, then any one of those enzymes should have a relatively similar effect when you remove it,” Dr. Finley points out. “What Ben noticed is that’s not actually the case.”
“The metabolic enzymes seemed to form two separate modules,” Jackson says. “This backed up the anecdotal evidence that we were accumulating that there were different parts of the TCA cycle that cells could use or not use.”
The CRISPR studies Jackson analyzed were performed in cancer cell lines — in other words, cells that aren’t “normal.” Arnold wanted to know if normal also engage in this alternative or noncanonical cycle. The Finley lab often works with embryonic stem cells, so Arnold had easy access to these normal cells. Arnold traced the flow of carbons through them and found that they also engaged in the noncanonical TCA cycle.
These two sets of experiments seemed to confirm that there really was an alternate way to perform the TCA cycle, one that is not in textbooks. But why had Krebs missed it?
To try to answer that question, Arnold decided to review Krebs’ original papers from the 1930s and 40s. She found, to her surprise, that Krebs had made his pivotal discoveries in one particular type of tissue: pigeon breast muscle.
“Nobody really talks about that,” Arnold says. “But it made us wonder if maybe different cell types have distinct preferences for whether they use the traditional TCA cycle or this alternate version.”
She decided to reconstruct Krebs’ original experiments, only in a dish rather than in pigeon muscle. She used mouse stem-like muscle cells to grow a muscle fiber precursor called a myotube and then traced the carbons. When she did this, she saw something interesting: “When the cells were still in a more stem-like stage, they seemed to be doing a lot of this noncanonical TCA cycle, similar to embryonic stem cells and cancer cells,” Arnold says. “But as soon as the cells had differentiated into myotubes, they immediately switched to the more traditional TCA cycle. This is in keeping with what Krebs saw in pigeon muscle tissue.”
To the team, this result suggested a clear link between changes in cell identity and usage of particular biochemical pathways. To test whether the changes in cell fate required use of the different pathways, the team performed additional experiments in which they chemically or genetically blocked certain enzymes in the cycles and asked whether the cells could still change their fate. They could not. This finding implied that changes in cell fate required different biochemical pathways.
To Burn or To Build
Why would a cell opt for a different form of the TCA cycle at all? According to Dr. Finley, the Krebs cycle is really good at maximizing ATP production. It helps cells combust all their nutrients down to carbon dioxide.
“That’s great if what you really care about is making ATP,” Dr. Finley says. “But if you want to grow, ATP is actually not the limiting reagent. You actually need to retain those carbons to make new biomass. That’s what the noncanonical TCA cycle does: It allows you to take carbons from glucose and export them to the cytosol, where they can be used to build other molecules. So, instead of burning the carbon, you get to keep it.”
This growth-oriented cycle may have particular relevance to cancer, whose signature characteristic is unlimited growth.
Dr. Finley cautions that their laboratory experiments were all done in a dish rather than in animals. The team is keenly interested in understanding whether and when it occurs in vivo, both in normal animals and in tumors.
“That will help us know whether it might be a good cancer drug target,” Dr. Finley says.
Join us in Targeting Mitochondria 2022 and stay updated about the most recent studies in cell biology. Keep in mind that professional speakers like Dr. Jessica Spinelli will boost your knowledge on the latest discoveries on the the mitochondria.
Media contact:
World Mitochondria Society
This email address is being protected from spambots. You need JavaScript enabled to view it.
+33-1-5504-7755
Targeting Mitochondria 2022 Congress
October 26-28, 2022 - Berlin, Germany
wms-site.com
Sustained Killing by Cytotoxic T cells: Mitochondrial Role
- Details
- Published on 18 March 2022
News Release, World Mitochondria Society, Berlin - Germany – March 18, 2022
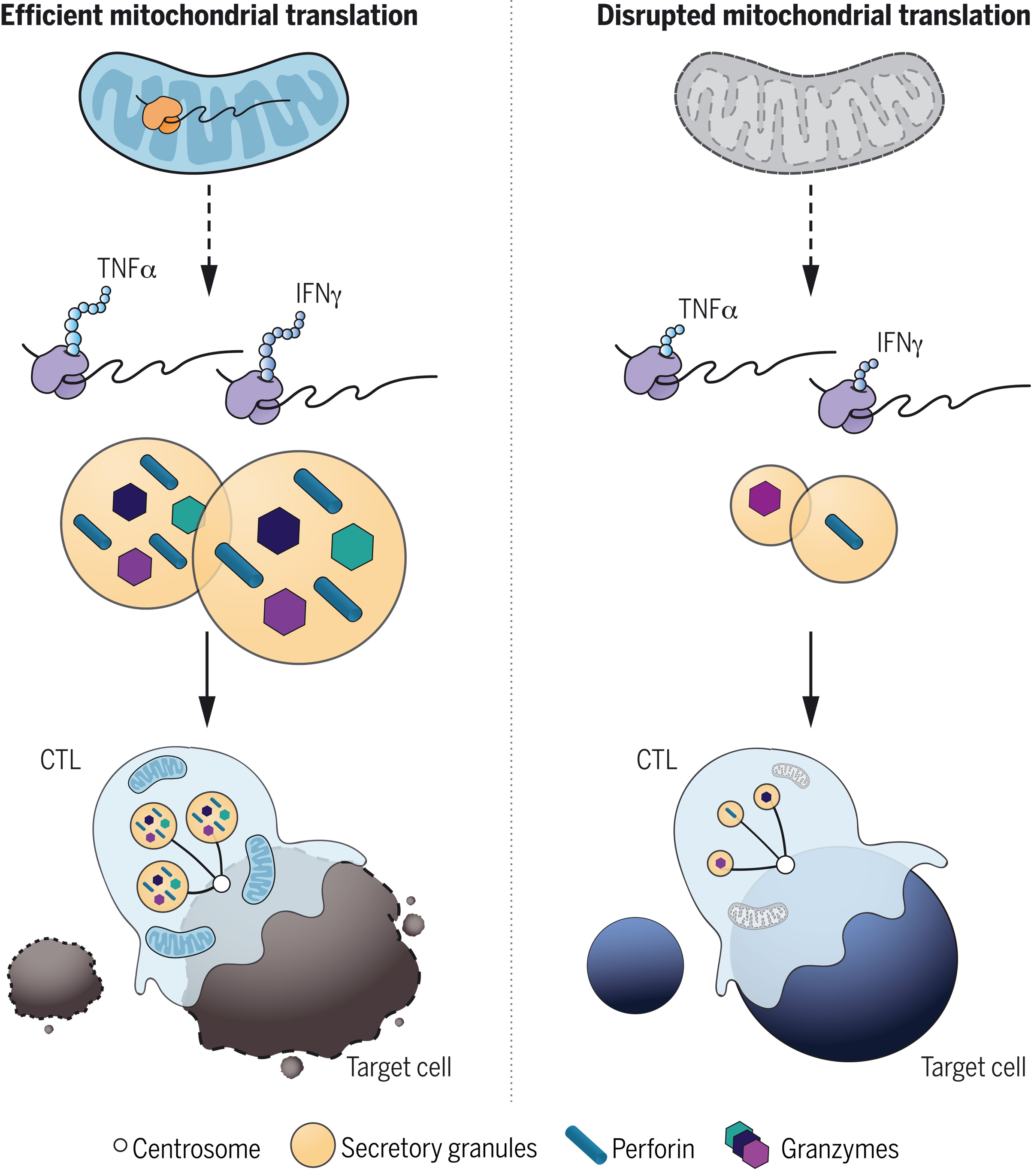
Cytotoxic T lymphocytes (CTLs) can terminate both virally infected cells and cancer cells by secreting cytolytic proteins such as perforin and granzyme B. Lisci et al. have identified mitochondria as important regulators of CTL killing; mice lacking the deubiquitinase USP30 have CTLs acutely depleted of mitochondria, and these cells have reduced killing ability but normal motility, signaling, and secretion.
Although mitochondrial mass has been correlated with CTL antitumor activity, CTLs show an increased reliance on glycolysis, suggesting a decreased dependence on mitochondrial respiration. Whether, how, or why mitochondria contribute as CTLs seek, recognize, and kill their targets is not well understood.
In this research, Lisci et al., generated CTLs from USP30-deficient mice to study the nature of this defect and to understand how it affects killing.
They reported the following:
T cell development was unaffected in USP30-deficient mice. However, upon activation, CD8+ T cells generated CTLs with an acute loss of mitochondria and impaired killing.
The cytotoxicity of USP30-deficient CTLs diminished with time, indicating a defect in sustained killing.
Despite the loss of mitochondria and decreased oxidative phosphorylation in Usp30−/− CTLs, motility, signaling, and secretion, which are required for killing, were all intact. However, the secretory granule size was reduced in Usp30−/− CTLs, with a reduction in newly synthesized intermediates of key cytolytic proteins, perforin and granzyme B. This suggested an underlying defect in the de novo protein synthesis, which is required for sustained killing.
Media contact:
World Mitochondria Society
This email address is being protected from spambots. You need JavaScript enabled to view it.
+33-1-5504-7755
Targeting Mitochondria 2022 Congress
October 26-28, 2022 - Berlin, Germany
wms-site.com
Mitochondrial Transfer in Obesity
- Details
- Published on 14 March 2022
News Release, World Mitochondria Society, Berlin - Germany – March 14, 2022
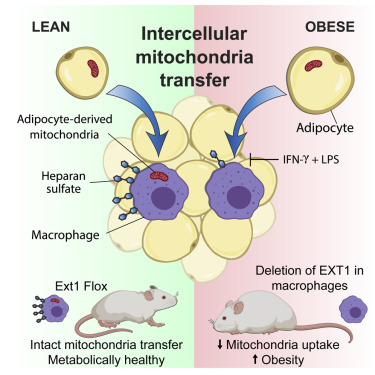
Researchers from the Brestoff and Teitelbaum Labs have demonstrated that adipose-tissue resident macrophages acquire mitochondria from adjacent adipocytes using Heparan Sulfate (HS). This process occurs in healthy conditions but is impaired in obesity. Further, they have shown that genetic disruption of mitochondria uptake by macrophages reduces energy expenditure and increases diet-induced obesity in mice, indicating that intercellular mitochondria transfer to macrophages mediates systemic metabolic homeostasis.
Obesity is an increasingly common metabolic disease that affects over 40% of adults and 18% of children and adolescents in the United States and is an independent risk factor for the development of many other disorders such as type 2 diabetes, cardiovascular diseases, and cancer.
Head of the group - Professor Jonathan Brestoff commented “Mitochondria are the power plants of cells, and it has long been assumed that they are made in one cell and never leave. We discovered that is not the case and found that fat cells give some of their mitochondria to an immune cell type called macrophages. In obesity, this transferring of mitochondria between cells goes awry, contributing to faster weight gain and worse metabolism. Using a tool called CRISPR, we screened the entire genome and figured out that cells trade mitochondria using a special type of sugar called heparan sulfate, which we think acts as a loading dock for receiving cargo like mitochondria. When we delete heparan sulfates on macrophages, mice get fat. This suggests to us that it is probably good for cells to trade mitochondria with each other. Our team is now trying to figure out how this mysterious and surprising process of mitochondria transfer works because we believe we can harness this biology to treat some human diseases.”
Dr. Wentong Jia, a postdoctoral fellow at the Brestoff lab added “The cell surface expression of heparan sulfate, a glycosaminoglycan required for mitochondria uptake in macrophages, depends on a key glycosyltransferase named EXT1. The 10E4 antibody from AMSBIO has helped us verify that we’ve successfully prevented Heparan Sulfate from being synthesized in cells that lack EXT1.”
“I find it fascinating that cells use heparan sulfates to take up mitochondria,” says Rocky Giwa, a Ph.D. candidate in the Brestoff Lab. “I wonder if there’s a correlation between the amount or composition of heparan sulfates and a cell’s ability to efficiently take up mitochondria from other cells. Since the various HS antibodies have unique specificities, the different clones can help us start to attack that question.”
Join us in Targeting Mitochondria 2022 where a whole session will be dedicated to Mitochondria Transplantation and Transfer; and professional researchers like, Dr. Camilla Bean, will introduce you to the importance of mitochondria in adipose tissue biology.
Media contact:
World Mitochondria Society
This email address is being protected from spambots. You need JavaScript enabled to view it.
+33-1-5504-7755
Targeting Mitochondria 2022 Congress
October 26-28, 2022 - Berlin, Germany
wms-site.com