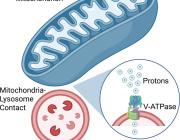
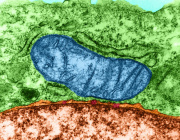
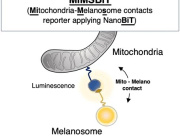
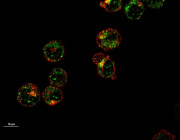
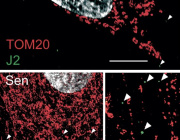
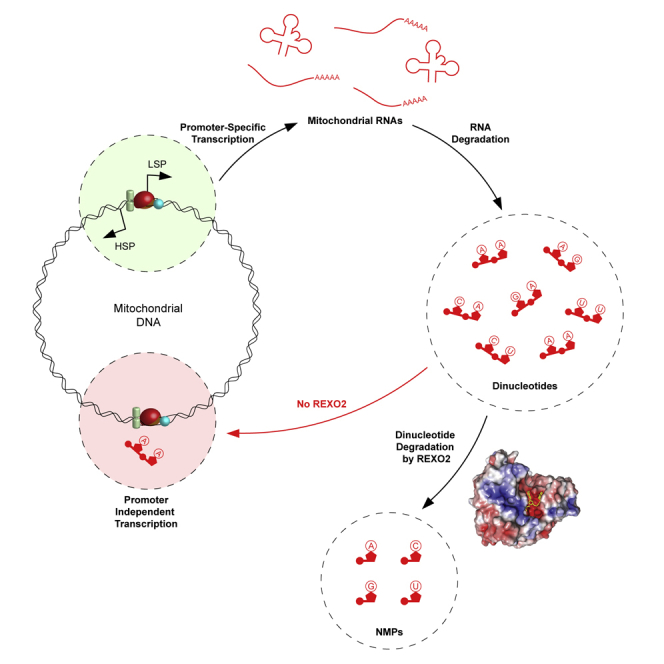
Mitochondrial RNA degradation is essential for life
- Details
- Published on 10 July 2020


Credit : pubmed
Researchers at Karolinska Institutet have discovered the essential role of the ribonuclease REXO2 in mitochondrial RNA degradation. The enzyme is essential for life, as a deficiency of it in mice has shown to be embryonic lethal. The study is published in the journal Molecular Cell.
We have asked two of the lead authors, Henrik Spåhr and Shan Jiang from the Nils-Göran Larsson group at the Department of Medical Biochemistry and Biophysics, about the most important results from their study.
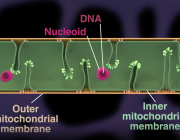
“The mitochondria are the power plants of our cells and are essential for converting the energy in the food we eat to a useful cellular energy currency. Importantly, mitochondria contain their own genetic material, mitochondrial DNA (mtDNA). MtDNA is totally distinct from the majority of our genetic material that is located in the nucleus. Expression of mtDNA is essential for the energy conversion of the cell, and the first step is to copy (transcribe) mtDNA to mitochondrial RNA, which, in turn, is the template for making 13 proteins that all are critically important for the cellular energy conversion. Quite a bit is known about how mitochondrial RNA is formed, but critical steps in the degradation have remained unknown. In the present study, we have identified an enzyme that is required for the last step of RNA degradation in human mitochondria, called REXO2”, Henrik Spåhr explains.
“This enzyme selectively degrades nanoRNAs (dinucleotides) and it is essential for life, as REXO2 deficiency results in embryonic lethality in mice.”
News source : www.news.ki.se
Authors : Nicholls TJ, Spåhr H, Jiang S, et al. Dinucleotide Degradation by REXO2 Maintains Promoter Specificity in Mammalian Mitochondria. Mol Cell. 2019;76(5):784-796.e6. doi:10.1016/j.molcel.2019.09.010
Enhanced axonal response of mitochondria to demyelination offers neuroprotection: implications for multiple sclerosis
- Details
- Published on 03 July 2020
 Credit: multiplesclerosisnewstoday.com
Credit: multiplesclerosisnewstoday.com
Axonal loss is the key pathological substrate of neurological disability in demyelinating disorders, including multiple sclerosis (MS). However, the consequences of demyelination on neuronal and axonal biology are poorly understood. The abundance of mitochondria in demyelinated axons in MS raises the possibility that increased mitochondrial content serves as a compensatory response to demyelination. Here, we show that upon demyelination mitochondria move from the neuronal cell body to the demyelinated axon, increasing axonal mitochondrial content, which we term the axonal response of mitochondria to demyelination (ARMD). However, following demyelination axons degenerate before the homeostatic ARMD reaches its peak. Enhancement of ARMD, by targeting mitochondrial biogenesis and mitochondrial transport from the cell body to axon, protects acutely demyelinated axons from degeneration. To determine the relevance of ARMD to disease state, we examined MS autopsy tissue and found a positive correlation between mitochondrial content in demyelinated dorsal column axons and cytochrome c oxidase (complex IV) deficiency in dorsal root ganglia (DRG) neuronal cell bodies. We experimentally demyelinated DRG neuron-specific complex IV deficient mice, as established disease models do not recapitulate complex IV deficiency in neurons, and found that these mice are able to demonstrate ARMD, despite the mitochondrial perturbation. Enhancement of mitochondrial dynamics in complex IV deficient neurons protects the axon upon demyelination. Consequently, increased mobilisation of mitochondria from the neuronal cell body to the axon is a novel neuroprotective strategy for the vulnerable, acutely demyelinated axon. We propose that promoting ARMD is likely to be a crucial preceding step for implementing potential regenerative strategies for demyelinating disorders.
Article Reference: Licht-Mayer, S., Campbell, G.R., Canizares, M. et al. Enhanced axonal response of mitochondria to demyelination offers neuroprotection: implications for multiple sclerosis. Acta Neuropathol (2020). https://doi.org/10.1007/s00401-020-02179-x
One-time treatment generates new neurons, eliminates Parkinson's disease in mice
- Details
- Published on 29 June 2020
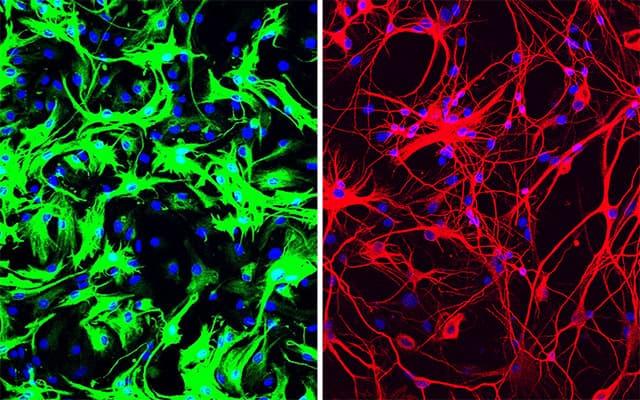 Left: mouse astrocytes (green) before reprogramming; Right: neurons (red) induced from mouse astrocytes after reprogramming with PTB antisense oligonucleotide treatment.
Left: mouse astrocytes (green) before reprogramming; Right: neurons (red) induced from mouse astrocytes after reprogramming with PTB antisense oligonucleotide treatment.
Credit: UC San Diego Health Sciences
Inhibiting a single gene converts many cell types directly into dopamine-producing neurons
Xiang-Dong Fu, PhD, has never been more excited about something in his entire career. He has long studied the basic biology of RNA, a genetic cousin of DNA, and the proteins that bind it. But a single discovery has launched Fu into a completely new field: neuroscience.
For decades, Fu and his team at University of California San Diego School of Medicine studied a protein called PTB, which is well known for binding RNA and influencing which genes are turned "on" or "off" in a cell. To study the role of a protein like PTB, scientists often manipulate cells to reduce the amount of that protein, and then watch to see what happens.
Several years ago, a postdoctoral researcher working in Fu's lab was taking that approach, using a technique called siRNA to silence the PTB gene in connective tissue cells known as fibroblasts. But it's a tedious process that needs to be performed over and over. He got tired of it and convinced Fu they should use a different technique to create a stable cell line that's permanently lacking PTB. At first, the postdoc complained about that too, because it made the cells grow so slowly.
But then he noticed something odd after a couple of weeks -- there were very few fibroblasts left. Almost the whole dish was instead filled with neurons.
In this serendipitous way, the team discovered that inhibiting or deleting just a single gene, the gene that encodes PTB, transforms several types of mouse cells directly into neurons.
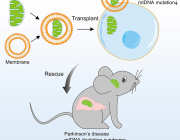
More recently, Fu and Hao Qian, PhD, another postdoctoral researcher in his lab, took the finding a big step forward, applying it in what could one day be a new therapeutic approach for Parkinson's disease and other neurodegenerative diseases. Just a single treatment to inhibit PTB in mice converted native astrocytes, star-shaped support cells of the brain, into neurons that produce the neurotransmitter dopamine. As a result, the mice's Parkinson's disease symptoms disappeared.
The study is published June 24, 2020 in Nature.
"Researchers around the world have tried many ways to generate neurons in the lab, using stem cells and other means, so we can study them better, as well as to use them to replace lost neurons in neurodegenerative diseases," said Fu, who is a Distinguished Professor in the Department of Cellular and Molecular Medicine at UC San Diego School of Medicine. "The fact that we could produce so many neurons in such a relatively easy way came as a big surprise."
There are several different ways to mimic Parkinson's disease in mice. In this case, the researchers applied a dopamine look-a-like molecule to poison neurons that produce dopamine. As a result, the mice lose dopamine-producing neurons and develop symptoms similar to Parkinson's disease, such as movement deficiencies.
The treatment works like this: The researchers developed a noninfectious virus that carries an antisense oligonucleotide sequence -- an artificial piece of DNA designed to specifically bind the RNA coding for PTB, thus degrading it, preventing it from being translated into a functional protein and stimulating neuron development.
Antisense oligonucleotides, also known as designer DNA drugs, are a proven approach for neurodegenerative and neuromuscular diseases -- study co-author, Don Cleveland, PhD, pioneered the technology, and it now forms the basis for a Food and Drug Administration (FDA)-approved therapy for spinal muscular atrophy and several other therapies currently in clinical trials. Cleveland is chair of the Department of Cellular and Molecular Medicine at UC San Diego School of Medicine and member of the Ludwig Institute for Cancer Research, San Diego.
The researchers administered the PTB antisense oligonucleotide treatment directly to the mouse's midbrain, which is responsible for regulating motor control and reward behaviors, and the part of the brain that typically loses dopamine-producing neurons in Parkinson's disease. A control group of mice received mock treatment with an empty virus or an irrelevant antisense sequence.
In the treated mice, a small subset of astrocytes converted to neurons, increasing the number of neurons by approximately 30 percent. Dopamine levels were restored to a level comparable to that in normal mice. What's more, the neurons grew and sent their processes into other parts of brain. There was no change in the control mice.
By two different measures of limb movement and response, the treated mice returned to normal within three months after a single treatment, and remained completely free from symptoms of Parkinson's disease for the rest of their lives. In contrast, the control mice showed no improvement.
"I was stunned at what I saw," said study co-author William Mobley, MD, PhD, Distinguished Professor of Neurosciences at UC San Diego School of Medicine. "This whole new strategy for treating neurodegeneration gives hope that it may be possible to help even those with advanced disease."
What is it about PTB that makes this work? "This protein is present in a lot of cells," Fu said. "But as neurons begin to develop from their precursors, it naturally disappears. What we've found is that forcing PTB to go away is the only signal a cell needs to turn on the genes needed to produce a neuron."
Of course, mice aren't people, he cautioned. The model the team used doesn't perfectly recapitulate all essential features of Parkinson's disease. But the study provides a proof of concept, Fu said.
Next, the team plans to optimize their methods and test the approach in mouse models that mimic Parkinson's disease through genetic changes. They have also patented the PTB antisense oligonucleotide treatment in order to move forward toward testing in humans.
"It's my dream to see this through to clinical trials, to test this approach as a treatment for Parkinson's disease, but also many other diseases where neurons are lost, such as Alzheimer's and Huntington's diseases and stroke," Fu said. "And dreaming even bigger -- what if we could target PTB to correct defects in other parts of the brain, to treat things like inherited brain defects?
"I intend to spend the rest of my career answering these questions."
News Source: www.eurekalert.org
Article reference:
Qian, H., Kang, X., Hu, J. et al. Reversing a model of Parkinson’s disease with in situ converted nigral neurons. Nature 582, 550–556 (2020). https://doi.org/10.1038/s41586-020-2388-4
The mitochondrial derived peptide humanin is a regulator of lifespan and healthspan
- Details
- Published on 29 June 2020
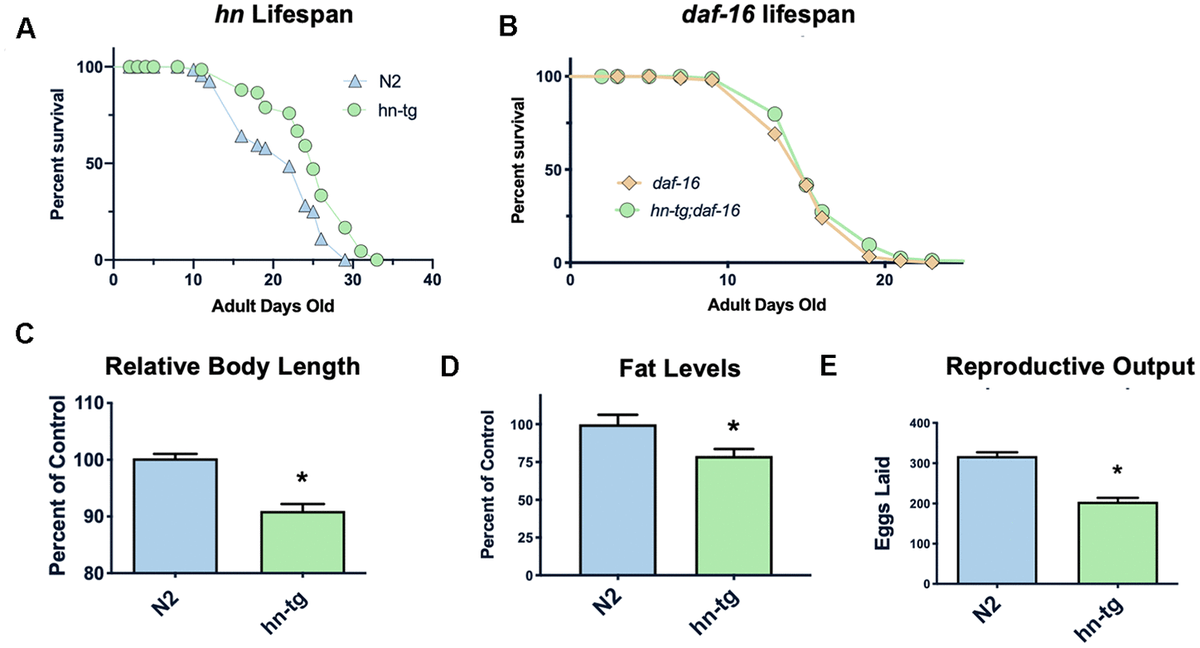 Figure 1. Humanin overexpression is sufficient to increase lifespan in C. elegans. Credit: aging-us.com
Figure 1. Humanin overexpression is sufficient to increase lifespan in C. elegans. Credit: aging-us.com
Humanin is a member of a new family of peptides that are encoded by short open reading frames within the mitochondrial genome. It is conserved in animals and is both neuroprotective and cytoprotective. Here we report that in C. elegans the overexpression of humanin is sufficient to increase lifespan, dependent on daf-16/Foxo. Humanin transgenic mice have many phenotypes that overlap with the worm phenotypes and, similar to exogenous humanin treatment, have increased protection against toxic insults. Treating middle-aged mice twice weekly with the potent humanin analogue HNG, humanin improves metabolic healthspan parameters and reduces inflammatory markers. In multiple species, humanin levels generally decline with age, but here we show that levels are surprisingly stable in the naked mole-rat, a model of negligible senescence. Furthermore, in children of centenarians, who are more likely to become centenarians themselves, circulating humanin levels are much greater than age-matched control subjects. Further linking humanin to healthspan, we observe that humanin levels are decreased in human diseases such as Alzheimer’s disease and MELAS (Mitochondrial Encephalopathy, Lactic Acidosis, and Stroke-like episodes). Together, these studies are the first to demonstrate that humanin is linked to improved healthspan and increased lifespan.
Article:
Unexpected insights into the dynamic structure of mitochondria
- Details
- Published on 20 February 2020

Credit: CC0 Public Domain
As power plants and energy stores, mitochondria are essential components of almost all cells in plants, fungi and animals. Until now, it has been assumed that these functions underlie a static structure of mitochondrial membranes. Researchers at the Heinrich Heine University Düsseldorf (HHU) and the University of California Los Angeles (UCLA), supported also by the Center for Advanced Imaging (CAi) of HHU, and have now discovered that the inner membranes of mitochondria are by no means static, but rather constantly change their structure every few seconds in living cells. This dynamic adaptation process further increases the performance of our cellular power plants. "In our opinion, this finding fundamentally changes the way our cellular power plants work and will probably change the textbooks," says Prof. Dr. Andreas Reichert, Institute of Biochemistry and Molecular Biology I at the HHU. The results are described in a publication in EMBO Reports.
Mitochondria are extremely important components in cells performing vital functions including the regulated conversion of energy from food into chemical energy in the form of ATP. ATP is the energy currency of cells and an adult human being produces (and consumes) approximately 75 kilograms of ATP per day. One molecule of ATP is produced about 20,000 times a day and then consumed again for energy utilization. This immense synthesis capacity takes place in the inner membrane of the mitochondria, which has numerous folds called cristae. It was previously assumed that a specific static structure of the cristae ensured the synthesis of ATP. Whether and to what extent cristae membranes are able to dynamically adapt or alter their structure in living cells and which proteins are required to do so, was unknown.
The research team of Prof. Dr. Andreas Reichert with Dr. Arun Kondadi and Dr. Ruchika Anand from the Institute of Biochemistry and Molecular Biology I of the HHU in collaboration with the research team of Prof. Dr. Orian Shirihai and Prof. Dr. Marc Liesa from UCLA (USA) succeeded for the first time in showing that cristae membranes in living cells continuously change their structure dynamically within seconds within mitochondria. This showed that the cristae membrane dynamics requires a recently identified protein complex, the MICOS complex. Malfunctions of the MICOS complex can lead to various serious diseases, such as Parkinson's disease and a form of mitochondrial encephalopathy with liver damage. After the identification of the first protein component of this complex (Fcj1/Mic60) about ten years ago by Prof. Andreas Reichert and his research group, this is another important step to elucidate the function of the MICOS complex.
"Our now published observations lead to the model that cristae, after membrane fission, can exist for a short time as isolated vesicles within mitochondria and then re-fuse with the inner membrane. This enables an optimal and extremely rapid adaptation to the energetic requirements in a cell," said Prof. Andreas Reichert.
News source: https://phys.org/news/
More information: Arun Kumar Kondadi et al, Cristae undergo continuous cycles of membrane remodelling in a MICOS ‐dependent manner, EMBO Reports (2020). DOI: 10.15252/embr.201949776